Gammaretrovirus vectors
Lentivirus vectors
Adenovirus vectors
Adeno-associated virus vectors
Herpes simplex virus-1 vectors
Cloning capacity
7–7.5 kb
18 kb
37 Kb
4.9 kb
Up to 152 kb (dependent on type of vector)
Transduction of non-diving cells
No
Yes
Yes
Yes
Yes
Integration into host chromosome
Yes
Yes
No
Yes
No
Immunological challenges
Few
Few
Triggers strong immune response
Few
Can induce an immune response (dependent on type of vector)
Pre-existing host immunity
Unlikely
Unlikely
Yes
Yes
Yes
Safety concerns
Insertional mutagenesis
Insertional mutagenesis
Inflammatory response/cytotoxicity
Inflammatory response, possible insertional mutagenesis
Cytopathic effects (dependent on type of vector)
Duration of transgene expression
Long term
Long term
Transient
Transient
Long term
Mode of entry into host cell
Fusion
Fusion
Receptor
Receptor
Fusion
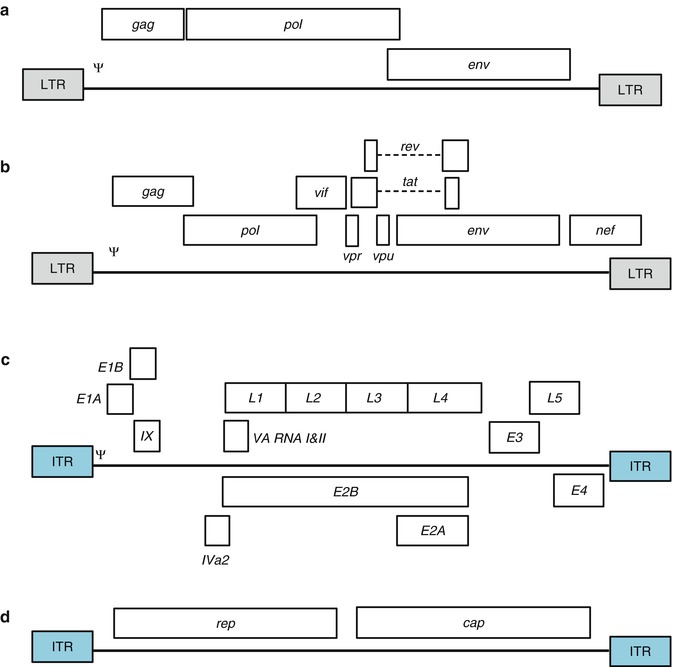
Fig. 7.1
Schematic of the genome organisation of the viruses commonly used to engineer viral vectors. Only the (a) Gammaretrovirus, (b) Lentivirus, (c) Adenovirus and (d) Adeno-associated virus genomes are shown and are not drawn to scale (Adapted from Giacca and Zacchigna [8], Verma and Weitzman [9]). Only the major genetic elements for each virus is shown: the long terminal repeats (LTRs) and inverted terminal repeats (ITRs) are boxed and shaded grey and blue, respectively. The packaging signal (Ψ) is also shown. See text for additional information
7.2.1 Retrovirus Vectors (RVVs)
Retroviruses (RVs) infect all vertebrates and those that infect humans are associated with various types of cancer, inflammatory diseases and human immune deficiency syndrome [12, 13]. This large family is classified into six genera that include alpha-, beta-, delta- and gammaretroviruses, lentiviruses and spumaviruses [14], which are based on genomic structure and sequence relationships. RVs are enveloped RNA viruses that consist of two identical copies of highly condensed positive-sense, single-stranded (ss) RNA enclosed by a capsid. The genome contains three essential genes, namely gag, pol and env that encode for the viral core proteins (matrix, capsid and nucleocapsid), the viral enzymes (protease, reverse transcriptase and integrase) and the viral envelope glycoproteins (surface and transmembrane proteins), respectively [9, 15–17]. RVVs efficiently transfer genes in vitro to a broad range of targeted cells and, since they have the capacity to integrate into the host genome, achieving long expression [2, 13, 18, 19]. For these reasons, RVs were among the first viruses engineered for gene therapy and have become the most commonly used RNA virus vectors [2]. Although an advantage, the integration of the vector into the host genome is also of major concern as random insertion into a pre-oncogenic site or a site responsible for the inactivation of tumor suppressor genes increases the risk of subsequent tumor development [10, 13, 20].
7.2.1.1 Gammaretrovirus Vectors (GRVVs)
Early retroviral systems were based on the gammaretrovirus Moloney-murine leukemia virus (Mo-MLV) [6, 21]. Mo-MLV vectors are integrative vectors that offer stable, long-term transgene expression, but a major limitation of using these vectors is that they function only in cells undergoing mitosis [2, 6]. Historically, MLV and other gammaretrovirus vectors were used to restore mutated gene functions in diseased cells [22] and introduce toxic [23] or suicide genes [24, 25] for therapeutic purposes. In the simplest examples of these vectors, the entire virus genome is deleted and replaced by the therapeutic transgene(s), with the exception of gag, pol and env flanked by the two viral LTRs; transcription of the transgene is directly controlled by the viral 5′ LTR [8].
7.2.1.2 Lentivirus Vectors (LVVs)
Lentivirus (LV) is a genus of the retrovirus family. Their genomes are more complicated than other retrovirus genomes, containing three to six additional accessory genes that regulate various stages of viral replication and contribute to the persistence of infection [2, 15, 16]. Since late 1990s, the use of LVVs for in vivo and ex vivo gene transfer applications has been studied extensively. Even though LVVs are mostly based on HIV-1, other LVs have also been used as vectors for gene therapy [26–31]. LVs have various properties that make them ideally suited as gene therapy vectors including their capacity to accommodate large or multiple transgenes [32]; the ability to maintain persistent gene expression by integrating the transgenes into the host cell genome; the absence of pre-existing anti-vector immunity in the host [33]; low anti-vector immunity in vivo [34, 35]; and limited potential for genotoxicity due to insertional mutagenesis [36, 37]. Also, due to the capacity of the LV pre-integration complex to actively cross the nuclear membrane [8], LVs do not require the breakdown of the nuclear membrane in order to integrate [38], thereby allowing efficient transfection of non-dividing cells [2, 26, 39, 40]. To date three generations of LVVs have been produced by deleting genes from the lentivirus genome, consistently improving efficiency and safety at each generation [2, 15, 41]. The current third generation replication-deficient HIV-1-based vectors contain only three (gag, pol, rev) of the nine HIV-1 genes [42], as well as the LTRs and the packaging signal [9]. This ensures that the stable integration of the provirus into the host genome does not result in the expression of any viral proteins, but only the expression of the transgene [43]. Furthermore, the development of self-inactivating LVVs improves their biosafety, minimizing mobilization of the LVV following infection with HIV [32]. Production of these vectors typically requires three or four plasmids, which include vector and packaging constructs [8, 15].
7.2.2 Poxvirus Vectors (PVVs)
The most extensively studied viral vectors are from the poxvirus (PV) family, a family of viruses that can infect both vertebrates and invertebrates [44, 45]. This family includes variants of vaccinia virus (from the Orthopoxvirus genus) as well as fowlpox and canarypox viruses (from the Avipoxvirus genus) [45]. PVs contain large linear dsDNA genomes. In contrast to other DNA viruses, poxviruses encode their own transcription machinery, a viral DNA-dependent RNA polymerase and post-transcriptional modifying enzymes, allowing self-sufficient virus replication in the host cell [44, 46]. Since poxvirus DNA is replicated in the cytoplasm, random insertion of the viral genome into the host chromosome is not a concern [18, 45, 47]. Additionally, PVVs have a wide host tropism, form stable recombinants, have accurate replication and efficient post-translational modification of transgenes and can accommodate large inserts of foreign DNA [44, 45]. Interestingly, some strains have a natural tropism for tumor tissue, with a 103–104-fold higher expression than in other organs [6]. PVVs have long been used in safe and successful vaccination programs, with the vaccinia vector used to vaccinate more than one billion people during the eradication of smallpox [44, 48–50]. To overcome safety concerns, attenuated vaccinia viruses (such as modified vaccinia virus Ankara or MVA) [51, 52], which can infect mammalian cells and express transgenes, but cannot produce infective virus particles, were developed [45, 53]. Unfortunately, vaccinia and MVA vectors can only be administered once or twice to vaccinia-immune or vaccinia-naïve patients due to the development of neutralizing antibodies to the vector [54].
7.2.3 Adenoviral Vectors (AdVVs)
Adenoviruses (AdVs) are non-enveloped DNA viruses than can infect and replicate in a wide range of organs, including the respiratory tract, the eye, bladder, gastrointestinal tract and liver [9]. Generally, these viruses are known to cause benign upper respiratory tract illness and epidemic gastroenteritis and conjunctivitis in humans. Human AdVs are classified into six species (A–F), which are further subdivided into more than 50 serotypes [1–10, 12–52] based on their ability to agglutinate red blood cells, oncogenic potential, genomic organization and DNA homology [2, 3, 55–57]. These viruses are produced and purified to high titres (up to 1013 virus particles/ml) and can infect both dividing and post-mitotic cells [6, 55], replicating very efficiently in permissive cells [5]. The AdV genome is easily manipulated and inserted transgenes are maintained throughout successive rounds of replication [5, 6]. To date, the vast majority of recombinant AdV vectors are derived from the human AdV serotypes 2 and 5 of species C [55, 58]. AdV vectors can be genetically engineered to remain replication-sufficient, conditionally replication-sufficient or replication-deficient in the host [58]. First generation vectors are generated by substituting the early gene 1 (E1), or the E1 and early gene 3 (E3), with an expression cassette [8, 58–61], rendering the AdV replication-deficient and capable of replication only in specifically designed complementing cell lines [55]. Second generation vectors have had the E1, E3, as well as early gene 2 (E2), deleted to minimize the host inflammatory response and potential toxicity [62], while third generation vectors (also known as gutted, gutless, high capacity or helper-dependent vectors) are generated by deleting the entire AdV genome with the exception of the ITRs (inverted terminal repeats) and Ψ (packaging signal) regions required in cis for viral DNA replication and packaging (see Fig. 7.1) [63, 64]. For this reason, a replication incompetent helper virus is required to provide all the necessary AdV functions in trans, which has to be later separated from the AdVVs [65]. Deletion of a large amount of genome increases the cloning capacity of the vector and the adaptive immune response of the host is also decreased [2].
7.2.4 Adeno-Associated Virus Vector (AAVVs)
Adeno-associated viruses (AAVs) belong to the Parvoviridae family containing viruses that infect numerous species of mammals, including humans [2, 7]. Even though more than 100 AAV serotypes have been identified, only 12 of these have been shown to infect humans, with AVV-2 the prototype [10]. AVVs are small, non-enveloped ssDNA viruses and the genome could consist of either the sense or anti-sense strand. The genome contains two genes (rep and cap which encode for seven proteins Rep40, Rep52, Rep68, Rep78, VP1, VP2, VP3), flanked by 5′ and 3′ palindromic sequences (ITRs). The Rep regulatory proteins are required for replication and packaging, whereas VIP1-3 are structural proteins that form the capsid; the ITRs, on the other hand, are indispensable for viral replication, packaging and integration. AAV requires co-infection with another helper virus (either HSV or an adenovirus) or a stressed-cell environment (e.g. when cells are irradiated or treated with genotoxic compounds) to mediate its replication; in the absence of these it establishes latency by integrating site-specifically into host chromosome 19 [2, 66–69]. The different AAV serotypes each use unique mechanisms for cell entry, which results in different host tropisms. A safe, non-rescueable helper plasmid is used to complement the AAV coding sequences for cap and rep in trans. Following infection of a permissible cell line by either a wild-type AdV or Herpes simplex virus (HSV), the two plasmids are co-transfected into the cells, allowing for the formation of recombinant AAV [9, 10, 70]. Alternatively, a helper virus-free procedure has been developed, in which a mini-adenovirus helper plasmid is co-transfected with the vector and a packaging plasmid into an AdV E1-expressing cell line [2, 9, 10], resulting in the absence of production of infectious AdV particles.
7.2.5 Herpes Simplex Virus Type 1 Vectors (HSVVs)
Wild-type herpes simplex virus-1 (HSV-1) is an enveloped, dsDNA virus that is spread by direct contact and infects and replicates in the skin and mucosal membranes, before infecting cells of the central nervous system [2, 10]. The large complex viral genome encodes for more than 80 proteins that can be classified as either essential or non-essential for virus replication [71, 72]. HSVVs mimic the latent state of HSV-1, producing a highly infectious, efficient vehicle for delivery of foreign genetic material to both neural and non-neural tissue cells [2]. Vectors engineered from HSV-1 have the capacity to deliver large pieces of foreign DNA (up to 150 kbs) to the nucleus of most dividing and non-dividing cells. HSV has several other advantages, including the fact that it can infect many different host cell types (including cells of the nervous system), the viral DNA will not integrate into the host genetic material, and the complex nature of the virus genome, which contains about 40 genes that are not essential for virus replication and can be deleted without interfering with virus production in vitro, while the latent behavior of HSV can be used for stable, long-term expression of therapeutic transgenes [11, 71, 73]. High titers of pure, non-pathogenic HSV-1 vectors can be produced by introducing null mutations into viral immediate early genes. This disrupts the capacity for viral replication, but allows production of the vectors by in vitro complementation of these genes in trans [2]. HSV-1 is currently genetically engineered to generate three different types of vectors i.e. (i) recombinant attenuated virus vector, (ii) defective, replication-incompetent non-pathogenic recombinant vector, and (iii) amplicon vectors. Of the HSVVs, amplicons have been used in most anti-cancer applications [11, 74, 75].
7.3 Non-viral Methods of Gene Delivery
The use of non-viral vectors (NVVs, natural or synthetic compounds) in which complexes of oligonucleotides, proteins, polymers or lipids are formed in particles capable of efficient transfer of genetic material into cells, precedes the development of viral vectors [10]. In the 1970s, the calcium phosphate-mediated transfection of cells was already widely accepted as an effective non-viral transfection tool [76]. In the same decade, Avery and colleagues reported phenotypic cellular changes following the non-viral delivery of exogenous DNA into pneumococcia [77]. Since those pioneering days, NVVs have been used to deliver synthetic oligonucleotide-based therapeutics that closely resemble traditional pharmaceuticals, including drugs, antibodies, RNA, as well as therapeutic genes to ‘diseased’ cells. Importantly, these vectors (i) are capable of being administered repeatedly to the host with little or no immune or inflammatory response, (ii) can be produced relatively easily in large quantities with high reproducibility and acceptable cost, (iii) are stable when stored at room temperature, (iv) can carry large transgene inserts, and (v) are easy to administer to patients [78–81]. In addition, non-viral vectors circumvent some of the problems associated with viral vectors including endogenous recombination, unexpected immune responses and oncogenicity [80, 81]. However, regardless of all the advantages of NVVs, they are generally rather inefficient at transfecting cells in vivo [6, 82, 83].
Plasmid DNA (pDNA) vaccines are synthesized from bacterial plasmids that have been engineered to express the therapeutic gene using promoter elements recognized by mammalian cells. The plasmids also contain a mammalian transcriptional terminator and a selectable marker to facilitate production of the plasmid in a bacterial system [84, 85]. Vaccination with pDNA mimics the natural intracellular pathogen gene expression pathways, activating both cellular and humoral responses. However, naked pDNA is generally difficult to deliver to the intended diseased site due to rapid clearance, degradation by cellular nucleases, the lack of organ-specific distribution and low efficiency in uptake after systemic delivery [86–89]. Even though nucleic acid modifications can overcome these shortcomings, specialized gene delivery vehicles that improve the delivery efficiency and cell-specificity, whilst protecting the pDNA against immune recognition are needed. Ideally, these gene delivery vehicles should also be able to enhance the therapeutic value of the transgene by providing complementary benefits such as the co-delivery of inflammatory suppressors to reduce potential cytokine production triggered by the naked pDNA [89].
7.3.1 Delivery of Plasmid DNA
7.3.1.1 Needle Injection
7.3.1.2 Liposome-Based Vectors
When a film of lipids is hydrated in an aqueous solution, it spontaneously forms microscopic particles consisting of one or more concentric lipid bilayers surrounding a watery compartment collectively called a liposome. These lipids consist of mono- or multi-cationic heads and a hydrophobic anchor held together by a linker [10, 91]. Cationic liposomes interact spontaneously with DNA to form lipoplexes [81, 92, 93]. Liposomes are the most effective non-viral vectors developed and offer several advantages for gene delivery to cells including, (i) low cost of synthesis and no resulting disease; (ii) DNA protection from degradation by cellular nucleases; (iii) transfer of large pieces of DNA; and (iv) being targeted to specific cells or tissues [10]. For these reasons, they are often the vehicle of choice for many applications [10, 89]. Even so, inflammatory toxicity, low transduction efficiency (compared to viral vectors) and immune recognition are problems still often associated with the use of liposomes as gene transfer tools [81, 89].
7.3.1.3 Polymer-Based Vectors
They are relatively easy to produce and flexible to modifications. These cationic polymers condense DNA into small particles, thereby preventing DNA degradation, ultimately improving gene expression. The DNA/polymer complex (or polyplex) is transferred into the cell via receptor-mediated endocytosis [81, 94]. Biodegradable polymers (known for their low toxicity and high biocompatibility) have been used to achieve controlled-release of DNA, further enhancing and prolonging gene expression. Controlled-release technology has been reported to increase and prolong the concentration of DNA around an injection site [80, 81], making it a very promising technology.
7.4 Cancer Gene Therapy
Since the first clinical gene therapy trial in 1990s, great attention has been paid to cancer as a potential candidate for gene therapy applications. Today, among a total of 1,902 gene therapy clinical trials worldwide, those for cancer gene therapy constitute about 64 %. This number not only represents the high enthusiasm in this field but also indicates the multiple pathways that can be targeted to stop cancer growth. However, the ongoing failure of any of these clinical trials to yield an efficacious gene therapy product, with the exception of the Chinese approved Gendicine, denotes the multiple challenges that are yet to be overcome. Several strategies have been attempted to stop cancer growth, for example through re- or over-expression of tumor suppressor genes [95], introduction of a suicide gene followed by pro-drug administration [96], knocking down oncogenes or enhancing radio-and chemo-sensitivity of the cancer cells [97]. Another strategy is aimed at targeting the host non-cancerous tissue, primarily to enhance the anti-tumor immune response. This can be achieved by expression of transgenes possessing direct anti-tumor activity (e.g. cytokines) or by indirectly activating the host immune response (e.g., GM-CSF) [98]. This last strategy can also convey chemo-protection to highly vulnerable cells in the bone marrow to protect them against frequent high doses of chemotherapy [99]. The inability to target all cancer cells, especially after tumor dissemination means that systemic cancer gene therapy aimed at developing anti-cancer immunity appear most promising. Specific targeting of cancer cells is also crucial as non-selective expression of toxic transgenes in normal cells can lead to severe toxicity. Fortunately, using promoters specifically upregulated in cancer cells to drive expression of suicide genes can be used, such as epidermal growth factor receptor promoter, transferrin promoter, telomerase promoter or Prostate Specific Antigen promoters [100, 101].
Some of the therapeutic targets used in cancer gene therapy are summarized below;
7.4.1 Oncogene and Tumor Suppressor Gene Targeted Gene Therapy
Normally cell cycle is controlled by two discrete types of signal; proto-oncogenes that promote survival and division as well as tumor suppressor genes that arrest the cell-cycle and initiate apoptotic programs. The interaction between these sets of genes determine the cell’s fate and any imbalance in gene expression can lead to malignant transformation.
Proto-oncogenes are genes that encode proteins responsible for stimulation of cell division, inhibition of cell differentiation and prevention of cell death. These genes are important for development and maintenance of human tissues and organs. However, their mutation is associated with neoplastic transformation [102]. These mutant proto-oncogenes are called oncogenes. Proto-oncogenes comprise three main classes; (i) growth factors and growth factor receptors; (ii) genes encoding proteins that work in the cellular cytoplasm such as; tyrosine and serine/ threonine kinases and (iii) nuclear transcription factors such as the NF-κB family [103]. Identification of these oncogenes provides a therapeutic platform for cancer treatment (Table 7.2). It is believed that silencing or knocking down these oncogenes can dampen cancer associated biological consequences at the gene level and thus treat or maintain the disease. Suppression of gene expression (gene silencing) can be achieved, to a variable degree, either at the transcriptional level by triplex-forming oligonucleotide or at the translational level using antisense RNA, siRNA, ribo-zymes and DNAzyme. Although targeting oncogenes with antisense RNA showed encouraging antitumor effects in vitro [104–106], human translation has not been easy. Many factors have to be considered including the gene expression profile of any tumor to determine which oncogenes are involved, as a single target will normally not be sufficient.
Table 7.2
Summary of ongoing gene therapy clinical trials in different types of cancer
Gene | Vector | Cancer type | Phase | Status | Trial ID |
|---|---|---|---|---|---|
Anti-oncogenes | |||||
TGF-β2 antisense | Plasmid DNA | Solid tumors | Phase I | Open | US-0908 |
Phase II | US-0393 | ||||
NSCLC | Phase III | US-0819 | |||
k-rasp53 antisense | Retrovirus | NSCLC | Phase I | US-0031 | |
c-fos or c-myc antisense | Retrovirus | MBC | US-0084 | ||
(EGFR) antisense | Lipofection | HNC | US-0285 | ||
Plasmid DNA | US-0576 | ||||
Tumor suppressor genes | |||||
P53 | Adenovirus | SCCHN | Phase I | Closed | BE-0003 |
NSCLC | Phase II | BE-0004 | |||
HNC | Phase II | BE-0007 | |||
Ovarian tumor | Phase II/III | BE-0008 | |||
HNC | Phase II | CH-0012 | |||
NSCLC | Phase II | CH-0013 | |||
NPC | Phase II/III | CN-0003 | |||
Phase III | Open | CN-0009 | |||
P53 | Adenovirus | Cervical cancer | Phase III | Open | CN-0010 |
Thyroid cancer | Phase IV | CN-0013 | |||
HNC | Phase III | Closed | DE-0037 | ||
Suicide genes | |||||
HSV-TK | Adenovirus | HCC | Phase II | Closed | CN-0011 |
Adenovirus | Recurrent HNC | Phase II | CN-0021 | ||
Retrovirus | Newly diagnosed previously untreated glioblastome | Phase III | BE-0002 | ||
Adenovirus | Prostate cancer | Phase III | Open | US-0842 | |
Tumor suppressor genes control cell growth in a dominant negative manner and their abnormal function relates to tumor development. These genes possess different functions to protect the host cell from cancer initiation and propagation. They are divided into three main groups; (i) caretaker genes such as the ataxia telangiectasia mutated (ATM) gene, which are responsible for maintaining genomic stability by ensuring the fidelity of DNA repair [107]; (ii) landscaper genes which act by maintaining or controlling the microenvironment harboring the growing cells [108]; and (iii) gatekeeper genes which provide a monitoring system to maintain the balance between cell division and death. Dysfunctional mutations of these genes lead to altered growth regulation and differentiation and predispose cells to tumor initiation and progression or metastasis [109].
Among the tumor suppressor genes, P53 gained the greatest attention because of its ability to monitor cellular stress, which could be induced by oncogene activation or DNA damage, and govern whether the cell will survive or enter the apoptosis pathway. The diversity of genes involved in cancer initiation often limits the effectiveness of gene therapy approaches. Fortunately, restoration or overexpression of single genes like P53 may be enough to induce tumor cell death. Various preclinical studies have demonstrated the efficacy of p53 in inducing apoptosis and suppressing tumor growth [110–112]. P53 based-gene therapy was also intensively tested in clinical trials (Table 7.2) with demonstrated safety and efficacy in some cancer types [113–116]. Moreover, P53 mediated delivery by adenovirus serotype 5 (Gendicine) has become the first approved gene therapy product, to treat carcinoma of the head and neck [117]. Conformational change mutants of P53 have also been reported in cancer cells. These changes lead to prolongation of the P53 life-span and augment its expression levels compared to normal cells, indicating that P53 could act as a tumor antigen [118], and targeting it with vaccine approaches may overcome the limitations associated with an inability to transfect all cancer cells. However clinical trials using virus-based vaccines encoding mutant P53 showed limited immunogenicity, attributed to the competition between the antigenicity of the vector and the P53 protein [119, 120].
7.4.2 Enhancing Pro-Drug Cytotoxicity in the Tumor Cells Through Gene Therapy
Confining the toxic effect of antitumor agents (such as chemotherapy and radiotherapy) to malignant cells remains a critical issue in the development of novel-cancer therapeutics. Alternatively, making cancer cells more sensitive to drug toxicity is possible through suicide gene therapy-based approaches. This involves enzymatically modifying cancer cells such that they convert an inactive pro-drug to an active toxic drug, hence increasing local concentrations of the toxic drug exclusively in the tumor and sparing normal tissues and organs. This can be achieved with several suicide gene therapy approaches.
7.4.3 Thymidine Kinase (TK)
Thymidine kinase (TK) is an ATP-thymidine 5′-phosphotransferase naturally present in all living cells. It is also present in viruses including herpes simplex virus (HSV), varicella virus, and Epstein-Barr virus. Unlike human TK, HSV-TK has the ability to phosphorylate some dNTP analogs such as Ganciclovir (GCV), a synthetic analogue of 2′-deoxy-guanosine. This integrates into newly synthesized DNA, terminating DNA synthesis and triggering the apoptotic signalling cascade. In the suicide gene therapy approach HSV-TK is delivered and expressed in cancer cells followed by systemic administration of GCV. Limitations in transduction efficiency are partially overcome by the bystander effect produced by the HSV-TK action (due to diffusion of the toxic metabolites to adjacent non-transduced cells) meaning there is no longer a necessity to achieve 100 % transduction efficiency within the tumor cells.
To date, several clinical studies utilizing HSV-TK suicide gene delivery systems (Table 7.2) to treat cancer have been reported, but with limited results [121–123]. The ability to transduce glioma cells in the brain with HSV-TK after single or multiple injections was demonstrated, however therapeutic effects were limited and associated with various degrees of toxicity [122]. A recombinant version of the suicide gene, HSV/TK-007 has been shown to be more effective than the wild-type gene product [124, 125]. Also co-administration of valproic acid, an inhibitor of histone deacetylases, enhances HSV-TK/GCV efficacy [126].
7.4.4 Cytosine Deaminase (CD)
A second gene that has been used to trigger selective destruction of tumor cells is the bacterial cytosine deaminase (CD), which encodes a protein capable of converting cytosine into uracil. This protein converts the nontoxic antifungal agent 5-fluorocytosine (5FC) into the wildly used toxic chemotherapeutic drug, 5-fluorouracil (5FU), which inhibits RNA and DNA synthesis during the S phase of the cell cycle [127]. This gene is not expressed in eukaryotic cells, therefore expression of CD gene in tumor cells leads to selective conversion of non-active 5FC to the toxic 5FU producing tumor-targeted chemotherapy. Therefore, the systemic side effects of 5FU are reduced and a local bystander effect is produced [128]. Unlike GCV, 5FC is a small, uncharged molecule that can pass freely in and out of the cell by diffusion meaning cell-cell contact is not required for a bystander effect [129]. Moreover it also produces a distant bystander effect through sensitization of the immune system [130, 131]. Initial clinical trials with the CD gene driven by a tumor specific promoter and injected into breast cancer patients using plasmid DNA showed high selectivity of expression in tumor cells, however minimal reduction in the tumor size were reported [96].
7.5 Anti-angiogenesis Gene Therapy
As tumors grow they require a supply of nutrients and oxygen designated as angiogenesis. No matter how unregulated, it is crucial for neoplastic growth, expansion and metastasis [132]. Several growth factors such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), platelet-derived growth factor (PDGF), epidermal growth factor (EGF), interleukins (ILs) and matrix proteins are mandatory for angiogenesis [133]. Moreover, proteolytic enzymes such as cathepsin, urokinase-type plasminogen activator, gelatinases A/B, and stromelysin are critical to the neovascularization process [134]. Anti-angiogenic modulators include natural inhibitors such as endostatin, thrombospondin and angiostatin or inhibitors of the pro-angiogenic factors (antibodies, antisense RNA and soluble receptors for FGF, VEGF). Therefore, anti-angiogenesis-based therapies targeting ECs instead of tumor cells are a potentially powerful therapeutic approach to fight cancer. They possesses several advantages including; reduced toxicity and less capacity to develop resistance. In addition, the therapy is independent on tumor type [135]. In addition, ECs in the tumor are more sensitive to anti-angiogenic therapy because they proliferate more rapidly and express several unique surface markers [136].
Nevertheless, most of the antitumor benefits reported in the clinic after targeting of angiogenesis were subtle and only achievable after intratumoral administration with very high doses of the agents. This could be explained by the presence of large numbers of factors that control tumor angiogenesis, as well as the malleability of cancer cells to compensate for any one missing factor. Therefore, as in most of the gene therapy approaches, combination of antitumor therapies is the most likely way to accomplish effective anticancer results with minimal side effects.
7.6 Cancer Vaccines and Vaccine Production
In recent years, substantial progress has been made in vaccine development for malignant diseases. New technologies have been developed for the identification of a large number of tumor antigens that can be utilized to stimulate the patient’s immune system in order to specifically recognize and destroy tumor cells. Cancer vaccination encompasses therapies that involve the administration of some form of antigen to induce a specific antitumor immune response. The vast majority of vaccine studies today employ measures designed to activate tumor-specific T cells. In addition to tumor antigens, cytokine genes (such as GM-CSF) or co-stimulatory genes (such as B7) are often included to improve the effectiveness of the vaccine. While the success of vaccine strategies are dependent on the route of antigen delivery and adjuvant, it is important to keep in mind that their success often depends as much on the particular antigen being used. This can be illustrated with the human mucin tumor antigen, MUC-1, which in early vaccine studies caused cellular immune responses in mice, but humoral responses in humans.
Related posts:
 Novel Strategy of Cancer Immunotherapy: Spiraling Up
Novel Strategy of Cancer Immunotherapy: Spiraling Up
 Psychoneuroendocrinoimmunotherapy of Cancer
Emerging Biomarkers During Clinical Development of Anti-CTLA4 Antibody Therapy
Psychoneuroendocrinoimmunotherapy of Cancer
Emerging Biomarkers During Clinical Development of Anti-CTLA4 Antibody Therapy
 Polarization of Tumor Milieu: Therapeutic Implications
Polarization of Tumor Milieu: Therapeutic Implications
 Photodynamic Therapy and Antitumor Immune Response
Photodynamic Therapy and Antitumor Immune Response
 Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


