Keywords
Suicide gene, GDEPT, cancer therapy, enzyme prodrug, thymidine kinase, nitroreductase, cytosine deaminase, carboxypeptidase G2, cytochrome P450, bystander effect
Introduction
Cancer chemotherapy has had an evolutionary history since the beginning of its modern period in the 20th century. Although the unsophisticated chemotherapy agents of the 1940s and 1950s, such as nitrogen mustard, elevated the hope for cancer treatment, their undesirable cytotoxic effects along with the limited remission of cancer and their uncontrollable “off-target” side effects intensified the search for more selective anticancer drugs.
Because of the cytotoxic nature of chemotherapeutic agents, finding a way to limit the lethal effect only to cancer cells and reduce the adverse effects on normal tissues has been of great interest. In 1957, Heidelberger et al. were the first to practically show the concept of “selective toxicity.” They showed that targeting the uracil uptake pathway is an efficient way of targeting tumor cells because the uracil pathway is more active in cancer cells than in normal cells .
In the 1970s, the emergence of monoclonal antibodies and later on their conjugations with cytotoxic agents started a new era of “targeted therapy” in cancer treatment . The main goal of targeted therapy is to exploit unique features of cancer cells, such as altered pathways and overexpressed receptors, to narrow down the toxic effects only to the tumor cells and reduce toxicity in normal tissues . In order to achieve this goal, a better knowledge of cancer biology and genetics is necessary. In the past decades, scientists have gained better insight about the signaling pathways involved in cell cycle and apoptosis, altered transcription, and metastasis mechanisms. Such discoveries have led to the identification of more target molecules for selective drug or gene delivery .
Among new emerging strategies for cancer gene therapy, one of the prominent ideas is the triggering of a self-destructive mechanism by transferring an exogene (transgene) to cancer cells. The product of the transgene might be naturally toxic for cancer cells (toxin gene therapy) or be a harmless enzyme that catalyzes the conversion of a nontoxic prodrug to a toxic compound . The latter approach is also known as gene-directed enzyme prodrug therapy (GDEPT) and is basically a two-step process. First, the enzyme-coding gene is selectively delivered to tumors followed by the systemic or intratumoral administration of a prodrug in a subtherapeutic dose . The accumulation of the prodrug’s toxic metabolites then causes target-specific death in the enzyme-bearing cells or the surrounding cells ( Figure 6.1 ). This method of gene therapy—also known as gene/prodrug activation therapy (GPAT) or suicide gene therapy (SGD)—aims to improve the therapeutic ratio (benefit versus toxic side effects) of cancer therapy .
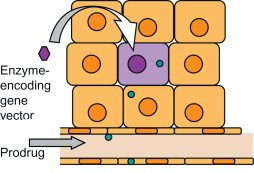
The two main factors that have a significant impact on the clinical success of GDEPT are the choice of enzyme and the choice of prodrug. Each year, a few new or modified enzyme/prodrug systems are introduced, and among them some find their way into preclinical and clinical studies. Later in this chapter, some of the most widely used enzyme/prodrug systems are discussed in detail, namely (1) herpes simplex virus thymidine kinase/ganciclovir (HSVTK/GCV), (2) cytosine deaminase/5-fluorocytosine (CD/5FC), (3) nitroreductase/CB1954 (NTR/CB1954), (4) carboxypeptidase G2/nitrogen mustard (CPG2/NM), (5) purine nucleoside phosphorylase/6-methylpurine deoxyriboside (PNP/MEP), and (6) cytochrome-P450/oxazaphosphorine (CYP450/Oxp).
One of the critical elements that govern the success of any enzyme/prodrug system is a potent bystander effect. Unlike other gene therapy systems in which only the cells that receive the therapeutic genes undergo apoptosis or necrosis, in GDEPT the therapeutic effect and cytotoxicity spreads from affected cells to neighboring cells as well. The following section discusses this phenomenon, which includes both bystander and distant bystander effects.
Bystander and Distant Bystander Effects
A bystander effect occurs when nontransduced or genetically unmodified cells are killed during death of transduced tumor cells ( Figure 6.2 ). This phenomenon was first suggested by Moolten and later elucidated by Culver et al . The bystander effect plays a crucial role in tumor regression because even under the best conditions, approximately 10% of tumor cells will be able to express the enzyme and consequently convert the nontoxic prodrug to its toxic metabolites . The problem of low transduction rate worsens even more in solid tumors, in which the necrotic layers at the core and debris at the outer part of tumors, poor angiogenesis, and disordered vascular pattern make the cancer cells at the inner layers inaccessible for gene delivery systems and prodrugs. This complexity has been reported to be one of the major reasons for the phase III clinical trial failure of the herpes simplex virus type I/ganciclovir GDEPT system . In this case, a 0.002% transduction rate was not sufficient enough to render a significant difference in tumor regression and survival time in patients treated with HSVtk/GCV as an adjunct therapy, especially because the HSV/GCV bystander effect is significantly affected by the presence of cell junctions. Perhaps other systems that act more independently from tumor physiology and microenvironment might be more beneficial .
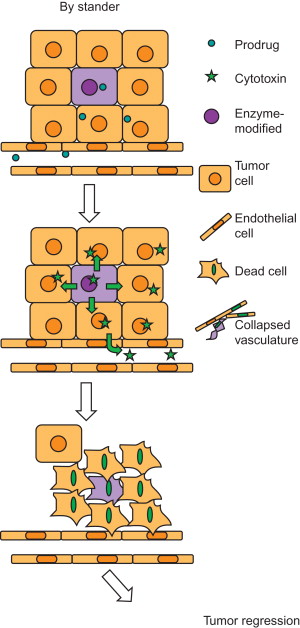
Several mechanisms are known to lead to bystander effect; of these, the predominant mechanism includes cell-to-cell diffusion of toxic metabolites from enzyme-expressing cells to neighboring cells either by passive diffusion from cell membrane or by active transport via intercellular connections (e.g., gap junctions) . Whereas neutral lipophilic drugs are capable of using passive diffusion, the highly charged molecules are transported through active diffusion . Highly charged metabolites such as ganciclovir triphosphate (GCV-TP) are dependent on gap junction intercellular communication (GJICs), which limits the success of GDEPT in vivo because in many cases the intercellular communications are distorted in cancer cells. To some extent, this explains the poor results obtained from preclinical and clinical studies on the HSVTK/GCV system. Evidence thus far points to the bystander effect being governed by cell-to-cell contacts rather than HSVTK expression level .
One of the main players in cell–cell intercellular communication is the connexin family, which encodes a family of transmembrane proteins known as GJIC proteins. Connexin expression in human cancers is in general reduced or undetectable. Several reports elucidate the positive relation between coexpression of one of the connexin family proteins (mostly CX26 and CX43) along with the enzyme (e.g., HSVTK) in augmenting the bystander effect .
The previously mentioned bystander effect is not the only mechanism mediating tumor regression in enzyme/prodrug gene therapy . The molecular mechanisms that lead to intratumoral bystander effect could also depend on an immune response in which tumor-infiltrating T cells might play a role . In 2001, Agard et al . reported a different kind of bystander effect, namely the “distant bystander effect,” in tumors that were anatomically not connected to the tumors that received gene therapy . In their study, colorectal xenograft tumors were formed in two distinct liver lobes of mice, and adenoviral vectors bearing HSVTK were injected into one of the lobes followed by intraperitoneal administration of GCV. In 46% of treated animals, the tumor in noninjected lobe also showed complete regression. Evidence showed the involvement of a cellular immune response as noted by the presence of both a CD8 + T cell infiltrate and an inflammatory reaction around the uninjected tumor lobe.
Rainov et al. also demonstrated an antitumor immune response in a number of patients with glioblastoma multiforme who had received HSVTK/ganciclovir gene therapy after surgery and radiotherapy . A significant increase in interleukin-12 (IL-12) and interferon-γ levels in patients who received gene therapy compared to the control group was detected. These findings are in accordance with other reports that confirm the activation of the immune system in response to antitumor antigens released in the blood after necrosis of cancer cells . These interesting observations indicate that a better understanding of the distant bystander effect mechanism can potentially improve therapeutic modalities in cancer.
The Enzyme/Prodrug Systems
The primary requirement for GDEPT to work involves a careful consideration of the employed enzyme and prodrug. For instance, the enzyme should not be toxic for the cells per se, it should possibly be expressed and activated in one step without the need for further post-translational modification, and its expression should not alter the cells’ housekeeping mechanisms .
In general, the enzymes that are used for GDEPT can be categorized into two groups: (1) exogenous with no counterpart in human and (2) endogenous with high expression in cancer cells and low expression in normal tissues . The nonhuman enzymes raise the possibility of immune response against the enzyme-expressing cells, especially if they are expressed for a long period of time . The immune response activation effect can be beneficial if it does not lead to the early removal of enzyme-expressing cells . The second group, although less likely to cause immunogenicity, may cause prodrug activation in off-target organs and consequent side effects . Because most of the human tumor cells lack tumor-specific enzymes that can be used as target the effectiveness of endogenous enzymes is still under investigation.
So far, a variety of techniques have been used to make enzymes more suitable for GDEPT. The aims of these techniques are to either divert the inherent affinity of enzyme from its natural substrate and increase its affinity toward the prodrug or improve enzyme characteristics such as enhanced stability and low immunogenicity to make it more suitable for use at the clinical level. In addition to the choice of enzyme, the prodrug is another important determinant in the success of suicide gene therapy. An ideal prodrug should be chemically stable with a half-life long enough to diffuse to neighboring cells but not too long to enter the systemic blood circulation. The prodrug should also show acceptable kinetics such as high affinity to the enzyme (low k m ) and high reaction rate (high K cat ). Preferably, it should be activated in one step and require no further modification by endogenous enzymes because in many cases the endogenous enzymes are severely modified or dysfunctional in cancer cells. Finally, it should be effective in both proliferative and quiescent cells .
Several enzyme/prodrug systems have been used for GDEPT. Table 6.1 provides a list of several known GDEPT systems, and next we discuss the most widely used ones along with their mechanisms of action.
| Name | Enzyme | Prodrugs | Drugs | Action |
|---|---|---|---|---|
| HSV-tk | Herpes simplex virus thymidine kinase | Ganciclovir | Ganciclovir monophosphate | Metabolized to triphosphate nucleotide; blocks DNA synthesis by inhibiting DNA polymerase |
| CD | Cytosine deaminase | 5-Fluorocytosine | 5-Fluorouracil (5-FU) | Blocks both DNA and RNA synthesis by inhibiting thymidylate synthetase |
| NTR | Nitroreductase | CB1954 and analogs | 5-(Aziridin 1-yl) 4-hydroxyl-amino 2-nitrobenzamide | DNA interstrand crosslinking agent; active in both dividing and nondividing cells |
| CPG2 | Carboxypeptidase G2 | Nitrogen mustard CMDA | Phenol-bis-iodo nitrogen mustard CMBA | DNA interstrand crosslinking agent |
| PNP | Purine nucleoside phosphorylase | 6-Methylpurine deoxyriboside | 6-methylpurine | Inhibits DNA, RNA and protein synthesis |
| CYP450 | Cytochrome P450 | Oxazaphosphorines: cyclophosphamide | 4-Hydroxycyclophosphamide | DNA interstrand crosslinking agent |
| CE | Carboxyesterase | Irinotecan (CPT11) | SN38 (camptothecin) | Causes DNA single strand breaks by binding to DNA topoisomerase I |
Herpes Simplex Virus Thymidine Kinase/Ganciclovir System
Unlike mammalian thymidine kinases, HSVTK exhibits a broad substrate specificity and a 1000-fold greater efficiency to selectively phosphorylate acyclovir-derived prodrug 9-([2-hydroxy-1-(hydroxymethyl) methyl) guanine, also known as ganciclovir (GCV). Frederic Moolten was the first to describe the potential of the HSVTK/GCV system as a prodrug-activating gene therapy . It was not until 1992 that HSVTK/GCV was tested as a gene therapy modality using retroviral vectors . Since then, HSVTK has become widely used as a suicide gene in combination with GCV for the treatment of a variety of cancers . The success of HSVTK/GCV therapy in cancer relies on the bystander effect, which has been studied extensively both in vitro and in vivo . It is based on the transfer of phosphorylated GCV from transduced to nontransduced cells mainly via gap junctions . Despite its broad use, the HSVTK/GCV system has several shortcomings, such as nonspecific toxicity due to high levels of both the transgene and GCV, relatively slow killing kinetics, and incomplete killing that is often due to the expression of inactive protein variants of HSVTK.
Attempts have been made to overcome the limitations of the HSVTK/GCV system by using novel HSVTK mutants. For example, protein engineering techniques such as site-directed mutagenesis have been used to modify enzyme kinetics and divert enzyme affinity ( K m ) from its natural substrate to the prodrug . The dose of GCV required for desirable effect causes severe side effects such as immune suppression. To minimize such adverse effects, random mutation studies are performed in order to identify the sequence of a construct with much higher specificity for GCV . Some mutants, particularly dm30-tk and SR39-TK, have shown increased specificity for GCV .
Studies have shown that the use of superior fusion enzymes such as mouse guanylate kinase (MGMK) with HSVTK offers significant advantages over wild-type HSVTK and HSVTK mutants alone, in at least two important ways: (1) by enhancing GCV-mediated cell killing and bystander killing effects and (2) by further reducing the amount of myelosuppressive GCV required for effective cell killing . One reason for the clinical success of HSVTK gene therapy is the ability to monitor therapy responses by using substrates suitable for positron emission tomography (PET) imaging . For instance, as shown by Alauddin et al . penciclovir has been modified to generate a radiolabeled analog 9-(4-[(18)F]fluoro-3-hydroxymethylbutyl)guanine (FHBG) for use in high-quality PET imaging studies . A phase II clinical trial is ongoing to investigate the suitability of [ 18 F] FHBG in breast cancer, glioma, and central nervous system (CNS) neoplasms ( http://clinicaltrials.gov , identifier NCT00185848).
In addition to modifications to HSVTK, ganciclovir analogs have also been introduced to circumvent shortcomings of GCV, such as poor bioavailability and toxicity. For example, whereas penciclovir, an ester analog of ganciclovir, and ganciclovir could both trigger apoptosis in HSVTK-expressing Chinese hamster ovary cells, penciclovir had reduced genotoxic effect and led to fewer chromosomal aberrations and recombination events. This makes penciclovir a better alternative to ganciclovir because it prevents neoplasia induced by chromosomal damage and rearrangements .
Cytosine Deaminase/5-Fluorocytosine System
The antimetabolite 5-fluorouracil (5FU) is widely used in the treatment of solid tumors, but its cytotoxicity to noncancerous tissues limits the dosage and frequency of drug administration. Cytosine deaminase (CD) is a pyrimidine salvage enzyme that catalyzes the deamination of cytosine to uracil. The bacterial or yeast CD converts an inert prodrug, 5-fluorocytosine (5FC), to a highly toxic chemotherapeutic agent, 5FU .5FU is a small molecule capable of diffusing into and out of cells, resulting in significant bystander effect . Several experimental studies have demonstrated that in addition to the 5FU-mediated bystander effect that does not require cell–cell contact, a distant bystander effect contributes to the success of this therapy . 5FU is both a chemotherapeutic agent and a radiosensitizer. Therefore, the combination of the CD/5FC system with ionizing radiation in in vitro and in vivo studies has shown better cell killing responses in comparison with GDEPT or radiation alone . Although in early studies, CD from Escherichia coli (bCD) was mostly used, it was later shown that the yeast cytosine deaminase (yCD) exhibits better kinetic properties for 5FC .
One of the limitations of the yCD/5FC system is the thermolabile nature of yCD at 37°C. To overcome this problem, two enzyme mutants (yCDdouble and yCDtriple) with increased half-lives and melting temperatures were identified that significantly increased the sensitivity of cancer cells to 5FC treatment compared with wild-type yCD . Leveille et al . showed the benefits of optimizing each step in a multicomponent therapy for efficient clinical outcome . They used a combination of CD::UPRT suicide gene expression and 5FC prodrug conversion, together with tumor-specific oncolytic vesicular stomatitis virus, to increase bystander killing. In addition, synergistic effects of a combination of CD/5FC and HSVTK/GCV, or double suicide gene therapy, have shown promise in preclinical studies . Jacobs et al . showed the use of CD as the therapeutic gene and HSVTK as a PET imaging reporter as well as a therapeutic gene to induce synergistic antitumor activity . In another study, Xing et al . monitored noninvasively suicide gene activity by tracking anabolism of 5FU to cytotoxic fluoronucleotides by magnetic resonance spectrometry in a rat prostate model . Phase I clinical trials are underway for high-grade gliomas, and phase II and III clinical trials are underway for replication-competent adenovirus-mediated suicide gene therapy, using double suicide gene therapy modality (yCD/5FC plus HSVTK/GCV) combined with intensity modulated radiation therapy for prostate cancer ( http://clinicaltrials.gov , identifier NTC00583492) .
Nitroreductase/CB1954 System
NTR/CB1954 is expected to demonstrate superior therapeutic efficacy because it kills both dividing and growth-arrested cancer cells, exhibits potent bystander effect, and lacks resistance to common chemotherapeutic agents such as cisplatin . CB1954 is a potent DNA alkylating agent that generates highly cytotoxic interstrand crosslinks in DNA . The NTR enzyme is responsible for reduction of nontoxic prodrug CB1954 to its activated cytotoxic form (5-(aziridin-1-yl)-4-hydroxylamino-2-nitrobenzamide) . CB1954 shows high selectivity (100- to 2000-fold) in a variety of NTR-transfected cell lines, including human ovarian (SKOV-3), colorectal (LS174T), and pancreatic (SUIT2, BxPC3) lines, with sensitivity correlating closely with the level of NTR enzyme expression. An additional immune bystander effect has also been observed whereby tumor cells killed by NTR/CB1954 can promote systemic immunity against the tumor . As mentioned previously, one significant advantage of the NTR/CB1954 system is that they do not discriminate between dividing and nondividing cells, but they require high prodrug dosages to obtain the desired effect resulting in side effects. To this end, several mutants of NTR have been generated and extensively studied . For example, Grohmann et al . developed a “mammalianized synthetic nitroreductase gene” that has been shown to improve sensitivity to CB1954 in mammalian cell lines . Recent studies have also attempted to mimic the dual capability of the HSVTK/GCV system with NTR as a therapeutic moiety fused to a reporter gene CytoCy5S, making it a promising strategy for future therapeutic evaluation of NTR in preclinical and clinical settings . Phase I clinical trials in prostate cancer of NTR in a replication-deficient adenoviral system with CB1954 have shown encouraging response .
Carboxypeptidase G2/Nitrogen Mustard System
CPG2 from Pseudomonas RS-16 is a 42-kDa exopeptidase in the aminoacylase-1/M20 family of enzymes that is not found in mammals. Although its natural substrate is folic acid, it also hydrolyzes glutamate from amidic, urethanic, and ureidic bonds. This makes it attractive medically as a rescue treatment for methotrexate overdose in addition to activating synthetic prodrugs . CPG2 catalyzes the hydrolysis of NM prodrugs, releasing glutamic acid and the cognate drug. The CPG2/NM system has several advantages over other GDEPT systems: (1) CPG2 has no mammalian equivalent, unlike carboxylesterase, purine nucleoside phosphorylase, or CYP450; (2) it has no additional activating steps, unlike CD/5FC or NTR/CB1954 systems ; and (3) gap junctions are not required for a bystander effect, unlike the HSVTK/GCV system, in which the activated drug (GCVTP) is charged and therefore cannot pass through cell membranes. CPG2-activated NM drugs are relatively lipophilic and can pass directly through cell membranes. The potential therapeutic effect of CPG2 in combination with prodrug 2 (4-[(2-chloroethyl 2-methylsulphonyloxyethyl) amino]benzoyl-l-glutamic acid) was first shown in antibody-directed enzyme/prodrug therapy . CPG2 plus another prodrug, 1(4-[bis(2-iodoethyl)amino]-phenyloxycarbonyl-l-glutamic acid), is the second antibody-directed enzyme/prodrug therapy system to reach clinical trials . The first report of CPG2-based GDEPT was that of Marais et al ., who tested the system in vitro using a range of human cancer cell types that had been transformed with the bacterial gene encoding CPG2 . A limitation of CPG2 was that the release of the enzyme from lysed cells could lead to generalized toxicity. CPG2 does not need cofactors and does not generate charged intermediate species. Marias et al . were able to show total kill of SKOV-3 cancer cells when only 12% of cells expressed CPG2. Other groups have also reported similar observations . Furthermore, surface tethering seems to be a determinant toward an increase in sensitivity to the drug and improved bystander activity . Stribbling et al . observed more than 90% of apoptotic cells from a population of cells in which only 10% expressed a mutant CPG2 by the surface tethering approach of CPG2 .
One major drawback associated with this system is significant immune system response to the enzyme due to its secretion into the bloodstream. To prevent this problem, CPG2 has been converted from secretory form to intracellular or cell membrane-anchored enzyme, resulting in significant reduction in immunogenicity . Throughout the years, structure–activity relationship studies have generated better prodrug candidates that improve the antitumor and bystander effects of the CPG2 system . Schepelmann et al . reported a targeted oncolytic virus-based CPG2 delivery system for colon carcinoma that not only efficiently targets the tumor cells but also exhibits excellent bystander effects .
Purine Nucleoside Phosphorylase/6-Methylpurine Deoxyriboside System
The E. coli PNP is a hexameric enzyme, catalyzing the glycosidic cleavage of purine ribonucleoside prodrugs such as 6-methylpurine 2-deoxyriboside (MEP-dr) and fludarabine (converted from fludarabine phosphate in plasma) to 2-deoxyribose-1-phosphate (or arabinose-1-phosphate) and free base compounds such as 6-methylpurine and 2-fluoroadenine, respectively. Both compounds are freely diffusible across cell membranes, allowing their spread from PNP transduced to untransduced cells, and are toxic to both proliferating and nonproliferating cells, thereby achieving a potent bystander effect . The bystander activity is facilitated by the nucleotide and nucleobase transporters across membranes in both directions and does not require cell–cell contact or gap junctions . Initial studies using prostate cancer cell lines and human hepatocellular carcinoma cell lines suggested that the PNP/MEP system is superior to the HSVTK/GCV system . Metabolites from PNP are incorporated during RNA synthesis and hence eventually block protein synthesis. Unlike GCV toxic metabolites, they are not dependent on tight junctions for diffusion to neighboring cells. Recently, a phase I clinical trial was started by PNP Therapeutics to investigate the safety of E. coli PNP/fludarabine phosphate in patients with head and neck cancers or other solid tumors ( http://clinicaltrials.gov , identifier NCT01310179).
Limitations due to immunogenicity from the bacterial origins of PNP led investigators to develop human PNP (hPNP) mutants that can cleave adenosine-based prodrugs that are not recognized by wild-type hPNP . These mutants are an attractive target because whereas the endogenous wild-type hPNP cannot use (deoxy)adenosine-based prodrugs as substrate, the mutants are highly effective at cleaving (deoxy)adenosine-based prodrugs and generating high levels of cytotoxic drugs in preclinical models . In human cells, adenine is salvaged from the extracellular environment by adenine phosphoribosyl transferase (APRT), which is responsible for the first and rate-limiting step in the activation of both MEP and F-Ade. Preclinical studies with PNP have shown that the enzyme activity from PNP transfected tumor cells is more than that of endogenous APRT, possibly causing MEP to diffuse away from the tumor. However, unlike the CD/5FC system in which UPRT plays a role in increasing sensitivity of the prodrugs, APRT overexpression does not improve the efficiency of the PNP system . In an effort to improve antitumor activity, designer nucleosides have been reported in combination with a structurally modified PNP enzyme . Unfortunately, this approach has not yet been successful in identifying prodrug/enzyme combinations that demonstrate better in vivo antitumor activity.
Cytochrome P450/Oxazaphosphorine System
CYP450 enzymes are a superfamily of heme-containing proteins that catalyze xenobiotic metabolism phase I reactions. The ability of CYP enzymes to bioconvert prodrugs to their active species could be exploited to a greater extent by taking advantage of the different CYP enzymes. This is done to achieve targeted drug delivery, improve efficacy, or decrease the unwanted adverse effects of existing and novel drug molecules. Although the different prodrug structures activated by CYP enzymes are broadly classified into (1) cyclic phosphates, phosphonates, and phosphoramides, (2) oximes, and (3) N -hydroxyguanidines, our focus is on the first category drugs, namely oxazaphosphorine drugs. The most prominent P450 GDEPT system employs the conversion of the oxazaphosphorine prodrugs ifosfamide (IFO) and cyclophosphamide (CPA) by human P450 isoforms into DNA-alkylating agents that cause cell death. The advantage of this system is locally increased concentrations of the active drug in the tumor coupled with reduced systemic toxicity due to additional conversion of prodrugs in the liver . Although both drugs have been used as DNA alkylating drugs in the treatment of various tumors for decades, severe toxicity (e.g., neurotoxicity, nephrotoxicity, and urotoxicity) associated with by-products released during their bioconversion has led to the development of safer CYP activated prodrugs. GDEPT exploits the enhanced expression of several distinct CYP forms in tumor cells for administering prodrugs to be activated within the malignant cells. In addition, a strong bystander effect that is not dependent on cell–cell contact but, rather, on drug diffusion effects accompanies the P450/IFO–CPA system, which adds to its overall therapeutic efficacy . Gunther et al . studied the effect of limited diffusion and low oxygen levels on increased bystander effect of the CYP21B/CPA system and showed that with approximately 25% CYP expressing cells, 80% cell killing could be achieved .
One of the potential problems associated with the CYP450 system is the ubiquitous expression of P450 reductase (P450R) that catalyzes P450 enzyme activity including cancer cells . The chemosensitivity of tumors to CYP450-activated prodrugs can be dramatically increased by transfer of a rodent or human CYP gene, which increases the capacity of the target tumor tissue to activate the prodrugs while decreasing immunogenicity . Another approach to enhance the efficiency of P450 gene delivery and P450 prodrug activation is by a replication-defective adenovirus armed with a CYP2B6-IRES-P450R expression cassette (Adeno-P450) that can sensitize human tumor cells of diverse tissue origin to CPA . The P450/CPA GDEPT approach has been shown to be safe and well tolerated in phase I clinical trials . In an effort to limit activation of the prodrug to the tumor, thereby minimizing the systemic effects of the activated prodrug, a combined GDEPT and cell-based therapy approach was taken by Kan et al. . This study used macrophages as a tumor-infiltrating carrier for a hypoxia-regulated GDEPT strategy based on the P450 and P450R genes to elicit bystander tumor cell killing in the presence of CPA. This approach not only diminished any undesirable adverse effects during treatment by withdrawing the prodrug but also allowed re-administration of genetically modified macrophages, should a repeat dose be required. This strategy was superior to the one based solely on adenoviral vectors in which preexisting anti-adenovirus antibodies formed following the first treatment. MetXia is a P450-GDEPT product candidate for use with CPA that showed good efficacy in phase I/II clinical trials in patients in the United Kingdom with advanced breast cancer or melanoma ( http://www.oxfordbiomedica.co.uk/page.asp?pageid=38 ). Furthermore, results from a phase I/II trial of MetXia in 35 patients with nonresectable pancreatic cancer have shown disease stabilization.
Targeted Cancer Suicide Gene Therapy
One of the major challenges of GDEPT is delivering the therapeutic genes to sufficient number of tumor cells and triggering a complete response. High selectivity of GDEPT is defined by how accurately the vector can deliver its cargo to the tumor environment (site targeting) or desired cells (entry targeting) or how specific the gene can be expressed in tumor cells (transcription targeting or post-entry targeting). Here, we discuss the main vector systems and recent achievements in terms of specificity in suicide gene therapy.
Entry Targeting: Vector-Mediated Suicide Gene Therapy
Both viral and nonviral vectors have been used for GDEPT. Because of the higher transfection efficiency, most of the vectors used for GDEPT studies are viral vectors, with adenoviral and retroviral vectors more abundantly used in preclinical and clinical cases. Use of other species such as vaccinia virus, adeno-associated virus, and herpes virus has also been reported .
Retroviral vectors are highly efficient transfection agents with low immunogenicity. Their tropism mostly depends on their envelope glycoproteins . Although integration of viral genome into host cell genome ensures the continuous high expression of transgene, the chance of insertional mutagenesis and its unknown consequences has led to the design of replication-deficient vectors . Although this design provides a level of safety, replication-deficient vectors were not very successful in in vivo studies because they cannot support a long-term gene expression . To overcome this problem, replication-competent retroviruses (RCRs) were designed, but their inability to controlled spread is still of major concern . RCRs are able to encapsulate their genome in tumor cells, so each infected cell will be a new virus-producing unit and the virus can infect more neighboring cells. Currently, a clinical trial is in progress to evaluate the safety and tolerability of the RCR vector-mediated suicide gene therapy in patients with recurrent high-grade glioma ( http://clinicaltrials.gov , NCT01156584).
Another virus type that has been widely used for GDEPT is adenovirus (Ad) . The internalization of adenoviruses takes place in a two-step process: (1) The interaction between adenovirus fiber knob and cellular primary receptor, coxsackievirus and adenovirus receptor (CAR), (2) followed by the interaction of penton base with the integrins on the cell surface. Unlike retroviruses, adenoviral vectors can infect both dividing and nondividing cells. Cell transduction is transient because the viral genome is unable to integrate into the host genome. The major drawback to the use of adenoviral vectors in GDEPT is their potential to evoke an immune response, especially after multidose injections . Similar to retroviruses, the first generation of adenoviral vectors lacked the ability to replicate in host cells. The replication-deficient adenoviral vectors provide a higher level of safety but decrease the efficiency of transduction. The next generation of adenoviral vectors, namely replication-competent adenoviral vectors, showed better results in terms of transduction levels .
Sitimagene ceradenovec (Cerepro) is the first adenoviral HSVTK vector to reach phase III clinical trials and is designed for eradication of residual cancerous cells after glioma surgery. Three clinical trials have been done to investigate the appropriate dose, safety, and efficacy of Cerepro . An ongoing phase III study using Cerepro with 250 patients has shown promising results .
A major drawback to the use of viral vectors is that they have a broad tropism, meaning they are incapable of cell-specific targeting. This drawback causes ex vivo and intratumoral protocols to be preferably chosen for viral GDEPT because systemic administration can cause severe side effects . In order to redirect the tropism of viruses to specifically target tumor cells, some attempts have been made to change the mechanism of virus internalization and create a “retargeted transduction” viral vector . For example, Morrison et al . used a hydrophilic polymer, N -(2-hydroxypropyl) methacrylamide (HPMA), to shield the viral capsid, followed by “recoating” of the virus by chemical conjugation of the murine epidermal growth factor . The new virus lost its infectious ability via CAR and was able to target an EGFR-expressing ovarian tumor cell line . In this approach, the post-internalization steps, such as virus unpackaging and propagation, were unaffected and comparable to those of unmodified virus-infected cells. Although CAR is expressed on most normal epithelial cells, data suggest that CAR expression may be highly variable in tumors, resulting in resistance to Ad infection . Consequently, modification to redirect adenoviral tropism to receptors highly expressed in cancer cells might improve tumor transduction and lead to an increased antitumoral effect. Huch et al . showed that in a pancreatic cancer model retargeting Ad-CYP2B1/CPA to fibroblast growth factor receptors resulted in a potent antitumoral effect and an increased survival rate, and it increased the potency of the CYP/CPA system .
As an alternative approach and to eliminate the deficiencies associated with the use of viral vectors, such as manufacturing difficulties, safety concerns, and constraints related to tissue targeting, synthetic nonviral vectors (e.g., cationic lipids and polymers) have been investigated. To date, one-third (~33%) of the vectors used in clinical trials have been nonviral vectors ( http://www.wiley.com.easyaccess2.lib.cuhk.edu.hk//legacy/wileychi/genmed/clinical ). Although nonviral vectors have lower transfection efficiency and hence express transgenes at a lower level, their safety and ease of scale up in pharmaceutical production and their higher capacity for exogenous DNA make them attractive for GDEPT. To date, liposomes, polymers, peptides, and naked DNA have been used for GDEPT, but only a few of them have reached clinical trials .
Cationic liposome–nucleic acid complexes (lipoplex) have been used in some in vitro and in vivo studies with relatively promising results. Before being recognized as a robust system for gene delivery, there are several weaknesses that need to be addressed. The reproducibility and stability of lipoplex production are reported to be low. In addition, some studies show that the transfection efficiency of lipoplexes appears to be nonreproducible in in vitro , in vivo , and ex vivo studies . These observations, especially with respect to in vivo experiments, may be due to the lipoplexes’ tendency to react with components of plasma and other body fluids and/or possibly being trapped in early endosomes or cytoplasm of the cells.
Recently, a transferrin/lipoplex system containing 1,2-dioleoyl-3-(trimethylammonium) propane (DOTAP)/cholesterol has been tested in vitro and in vivo . The HSVTK gene was transferred to oral cancer cells in culture, using DOTAP/cholesterol polyplexes equipped with transferrin as a targeting moiety for dividing and nondividing cells. After adding GCV, 100% toxicity due to apoptosis induction was reported. The same combination was used in a murine model of head and neck squamous cell carcinoma with HSVTK/GCV and yCD/5FC as a dual suicide gene therapy . A significant decrease in tumor size (approximately three- to fivefold) was observed in lipoplex-mediated pCMV-TK/GCV therapy. No difference between HSVTK/GCV and yCD/5FC in terms of tumor regression was reported in this study. An immune response was also reported in animals receiving DOTAP/HSVTK/GCV as indicated by infiltration of CD8 + T cells, natural killer cells, and an increase in IL-2 and inflammatory cytokine levels. The immune stimulating effect was reported to be related to CPG island on DNA or the cationic lipids .
In a study by Wang et al . a multifunctional recombinant fusion vector was used for targeted delivery of SR39 (HSVTK mutant) to HER2-positive SKOV-3 cancer cells . The fusion vector was equipped with a HER2 targeting affibody, a DNA condensing motif derived from human histone H2A, and the fusogenic peptide GALA . The vector in combination with SR39 and GCV showed 80% toxicity to SKOV-3 cancer cells in vitro. In a nude mouse model, the vector in complex with SR39 and in combination with GCV was able to significantly inhibit tumor growth, and the effects were comparable to the effects caused by Ad/SR39 .
Post-Entry Targeting: Tumor-Specific Promoter-Mediated Cancer Cell Targeting
The specific expression of an enzyme in a defined tumor cell type is the fundamental basis for the success of GDEPT. Another method to increase the specificity of GDEPT and reduce the risk of off-target adverse effects is transcriptional targeting or “post-entry targeting,” which brings the expression of transgene under the control of a tumor-specific promoter. A few such promoters are discussed here.
Carcinoembryonic Antigen
The carcinoembryonic antigen (CEA) promoter is a fetal promoter that is reactivated in many carcinomas. This promoter has been assessed in a number of suicide gene therapies designed to treat colorectal carcinoma . In one study, it was shown that a decrease in expression level (10- to 300-fold) occurred when the expression of yCD was mediated by CEA instead of cytomegalovirus (CMV) or Rous sarcoma virus (RSV) promoter . To make up for such a dramatic decrease in expression, an enhancer sequence upstream of the CEA promoter was utilized. As a result, the in vivo expression level was augmented by 30-fold compared to the basal CEA-mediated yCD expression, and this was comparable to RSV-induced expression .
Hypoxia-Responsive Elements
Another approach for transcriptional targeting is based on the special tumor micromilieu. Poor vascularization and high rate of metabolism usually cause hypoxia in solid tumors. A number of studies on erythropoietin-producing cells noted that in response to hypoxia, many pathways are initiated in cells. The starting point is usually binding of hypoxia-inducible factor to hypoxia response element (HRE), which is an enhancer sequence . By constructing a series of synthetic promoters consisting of HRE of vascular endothelial growth factor (VEGF) gene or erythropoietin gene and their combination with minimal CMV promoter (mCMV) or minimal interleukin-2 (mIL-2) promoter, hypoxia-dependent expression of either of two suicide genes, HSVTK and/or bacterial nitroreductase (NTR), in various cancer cell lines was induced .
Radiation-Responsive Elements
Radiation is a well-established single or combination therapy for solid tumors and has often been used for transgene activation in tumor cells. In combination therapy, when used as an activator, a smaller dose of radiation is needed. Reactive oxygen species (ROS), produced by radiation, can activate the early growth response (Egr)-1 gene, and it has been shown that some elements in (Egr)-1 promoter, namely CArG, respond directly to ROS . In parts of tumor with low oxygenation, the production of ROS is disrupted or ceased. Therefore, the cells in hypoxic areas of tumor show resistance to radiation or radiation-induced gene therapy. In order to combine hypoxia and radiation-sensitive elements, a promoter was synthesized by combining tandem repeats of HRE and CArG as the enhancers for CMV basal promoter. This complex was proven to react to hypoxia, radiation, or both . The aforementioned promoter controlled the expression of a gene encoding the Cre recombinase enzyme. After being exposed to stimuli, the expressed Cre recombinase recognized a stop cassette located in CMV promoter sequence, flanked by two loxP sequences. Subsequently, the stop cassette was removed by the Cre recombinase, and the intact CMV promoter mediated the expression of the HSVTK gene. This elegant system for enhancing expression levels showed an efficient lethal activity in vivo and tumor regression in 71% of human glioma xenograft models .
Human Telomerase Reverse Transcriptase
Another specific feature that has been exploited for GDEPT is overactivation of the telomerase enzyme in cancer cells. Except for peripheral blood leukocytes and special stem cells, telomerase activity is almost absent in many somatic cells. The expression of the catalytic subunit of telomerase is controlled by the human telomerase reverse transcriptase (hTERT) promoter, which has been used widely for cancer virotherapy and gene therapy . Systemic injection of an armed oncolytic adenoviral vector in which the expression of carboxypeptidase enzyme was mediated by hTERT showed apoptosis induction in human colon carcinoma cell lines and xenografts but not in surrounding normal tissue . A phase I clinical trial for intra-abdominal cancers such as ovarian, colon, pancreatic, and gastric is underway for Ad-hTR-NTR, an adenoviral vector with nitroreductase and hTR, another promoter of the telomerase subunit .
Site Targeting: Stem Cell-Mediated Tumor Targeting
Discovering the inherent tropism of stem and progenitor cells for solid tumors and their ability to migrate to tumor loci and infiltrate and participate in stroma formation around the tumor was one of the milestones in cancer gene therapy, especially for inaccessible or difficult-to-access solid tumors such as glioblastoma. The hallmark of cell-based therapy is its ability to pass the blood–brain barrier and, in some cases, engraft to the tumor or migrate to disseminated tumors in different anatomical locations—an ability that is rare when using nonviral and viral vectors .
The targeting ability of modified viruses and nonviral systems is highly dependent on the expression of a specific single receptor or biomarker on cancer cells. Because each tumor is a heterogeneous collection of cells with different origins, the targeted biomarker might not be present in all tumor cells or might be downregulated or altered in different tumor microenvironments. The tropism of stem cells, on the other hand, is a reaction to a density gradient of cytokines and pro-angiogenic growth factors produced in a tumor site and is independent of the expression of any specific protein . Another advantage of using primary stem/progenitor cells is their negligible immunogenicity. Hori et al . examined the potential immunogenicity of neural stem cells . They noted that progenitor cells isolated from mammalian CNS do not express histocompatibility complex (MHC) I and II in vitro . Even 4 weeks after injection beneath the kidney capsule in vivo , neither expression of MHC I/II nor host sensitization were detected .
Based on these positive features, many in vitro and in vivo studies have exploited stem cells as vectors for GDEPT. So far, stem cells from neural, adipose tissue, and bone marrow have been recognized to be beneficial for preclinical trials. Neural stem cells (NSCs) are CNS progenitor cells with the ability to differentiate to all three kinds of CNS cells. They have been used widely for the treatment of different tumors, such as breast cancer, melanoma brain metastases, and intracerebral medulloblastomas, although their main application is in difficult-to-access brain tumors .
The first step in stem cell-based gene therapy is to engineer them to produce the prodrug converting enzyme. Depending on whether transient or stable expression is needed, different viral vectors have been employed. Whereas retroviral and lentiviral vectors provide a stable expression by integrating into the host genome, adenoviral vectors mediate a high level of transient expression . Baclovirus-based vectors that can transduce both dividing and nondividing cells have been reported to stably express HSVTK even 3 weeks after systemic injection of NSCs derived from human embryonic stem cells .
There are many NSC-based GDEPT studies, most of which have used HSVTK/GCV or yCD/5FC as their enzyme/prodrug systems . In a recent study, Cho et al . engineered yCD expressing human NSCs and used them for the treatment of human glioma in a rat model. Systemic administration of 5FC along with injection of NSCs caused not only a significant tumor regression (47 and 67%, respectively, for intratumoral and systemic administration) but also a significant increase in survival rate . NSCs expressing yCD also led to a reduction of brain tumor size in mouse models, triggering a mild immune response and macrophage infiltration to the tumor site, which was suggested to improve the tumor regression process . Other enzyme/prodrug systems have also been used successfully in stem cell-mediated GDEPT. For example, Danks et al . engineered neural stem cells with the ability to express rabbit carboxylesterase enzyme (rCE). After intravenous injection, these cells migrated to disseminated neuroblastoma in liver and bone marrow, and upon the administration of prodrug CPT-11, long-term survival in a mouse model was observed . In another study, engineered mesenchymal stem cells from adipose tissue expressing CD::UPRT enzyme were successfully used for prostate tumor regression both in vitro and in vivo . In a preclinical setting, the intravenous injection of these cells along with systemic administration of 5FC showed total prevention of tumor establishment when they were injected concomitantly with human bone metastatic PC cells or a significant decrease in tumor size when they were injected intravenously to animal models bearing prostate tumors . A pilot clinical study is ongoing to investigate the feasibility of engineered NSCs expressing bacterial CD and oral 5FC in patients with high-grade gliomas ( http://clinicaltrials.gov , NCT01172964).
The use of nonviral vectors for in vitro engineering of stem cells seems to be restricted in stem cell-mediated prodrug gene therapy mostly because of their low efficiency compared to their viral counterparts. However, their safety and ease of production encourage their use in the stem cell transduction process, especially because some reports indicate that the immune response to stem cells might not be against the stem cells per se but also due to the viral vectors used for transduction of transgene . Cationic lipoplexes, liposomes, and other nonviral vectors have been used for stem cell transduction, although the engineered stem cells have not been used directly for GDEPT .
Related posts:
 Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
 Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy
Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy
 Clinical Trials Using LV-P140K-MGMT for Gliomas
Clinical Trials Using LV-P140K-MGMT for Gliomas
 Therapeutic Efficacy and Systemic Antitumor T Cell Immunity Induced by RheoSwitch-Regulated IL-12 Expression after Intratumoral Injection of Adenovirus Vector or Vector-Transduced Dendritic Cells
Therapeutic Efficacy and Systemic Antitumor T Cell Immunity Induced by RheoSwitch-Regulated IL-12 Expression after Intratumoral Injection of Adenovirus Vector or Vector-Transduced Dendritic Cells
 Targeting Tumor Vasculature Using Adeno-Associated Virus Phage Vectors Coding Tumor Necrosis Factor-α
Targeting Tumor Vasculature Using Adeno-Associated Virus Phage Vectors Coding Tumor Necrosis Factor-α
 Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


