Mutation-activated signaling from the KIT and PDGFRA kinases has been successfully targeted in gastrointestinal stromal tumors (GISTs), with subtle differences between the mutations serving to refine prognosis and more precisely tailor therapy. There is a growing understanding of the molecular drivers of GISTs lacking mutations in KIT or PDGFRA, so called wild-type GISTs, further aiding in management decisions. This article provides an overview of all the known molecular subtypes of GIST and provides information about clinical correlates, treatment, and prognosis depending on the subtype.
Key points
- •
Recent advances in the knowledge of gastrointestinal stromal tumors (GISTs) have been translated into improved diagnosis and treatment.
- •
The current understanding is that GISTs represent a heterogeneous collection of molecular entities linked by a common histology and presumed cell of origin.
- •
Most GISTs are driven by a pathogenic mutant kinase, but other disease-initiating molecular abnormalities have been identified.
- •
The type of underlying molecular defect in a given patient’s GIST has a significant impact on treatment response and potential mechanisms of primary and secondary resistance.
Gastrointestinal stromal tumors (GISTs) were not widely recognized before 1998. They are now regarded as the most common mesenchymal tumor of the gastrointestinal (GI) tract, with an incidence of more than 5000 cases per year in the United States. These tumors have become a paradigm for the expanding use of gene-based diagnostics and molecularly targeted therapies. This review focuses on recent advances in our understanding of GIST biology and how this knowledge has been translated into improved diagnosis and treatment. In addition, the evolving molecular classification of GIST is updated, with particular emphasis on those 10% to 15% of GISTs that lack gain-of-function KIT or platelet-derived growth factor receptor α (PDGFRA) receptor tyrosine kinase (RTK) mutations. These GISTs were previously classified as wild-type (WT) GIST, but we now propose the use of the term RTK-WT as a more accurate subclassification of this group of GISTs.
Pathology and diagnosis
GISTs most commonly arise in the stomach (60%) but also present in the small intestine (25%), rectum (5%), and other sites along the GI tract, including the esophagus, colon, appendix, and gallbladder. Rarely, the tumors appear unattached from the GI tract. These so-called extraintestinal GISTs may occupy the mesentery or omentum. GISTs are rare outside the abdominal cavity, but there is a well-documented report of a primary GIST of the pleura.
Historically, GISTs were considered to be sarcomas of smooth muscle origin, because of their predominantly spindle cell morphology and their association with the muscularis propria. However, studies using electron microscopy and immunohistochemistry suggested that these tumors differ from classic leiomyosarcoma, and in 1983, Mazur and Clark proposed the term stromal tumor. The subsequent discovery that most stromal tumors arising in the GI tract were CD34-positive provided further support for their distinction from leiomyosarcoma.
During the 1990s, several investigators noted similarities between GISTs and a little known population of cells in the gut wall designated as the interstitial cells of Cajal (ICCs). ICCs are now known to serve as pacemakers for peristaltic gut contractions. Studies during this period showed that ICCs express KIT tyrosine kinase (CD117) and are developmentally dependent on stem cell factor (SCF) signaling through this kinase. This finding led to the observation by 2 different groups in 1998 that GISTs commonly express CD117. It is now well established that 95% of GISTs are immunohistochemically positive for CD117.
In 2004, DOG-1 (also known as anoctamin 1) was described as another high-specific marker for GISTs. DOG-1 is a calcium-activated chloride channel that is highly expressed in ICCs and is detectable in 98% of GISTs, regardless of CD117 expression. Only a small percentage of other sarcomas stain positively for DOG-1. The combination of CD117 and DOG-1 expression is essentially diagnostic for GIST.
The use of CD117 and DOG-1 has helped define the range of morphologies associated with GIST. Although most GISTs are composed of a uniform population of spindled cells, some have an epithelioid appearance and others comprise a mixture of spindled and epithelioid cells. Tumor cellularity varies widely among GISTs. Low-grade lesions may show areas of central calcification, or show a bandlike alignment of nuclei that mimics a schwannoma. High-grade tumors often ulcerate the overlying mucosa and may undergo significant hemorrhagic necrosis. The variety in GIST histology dictates a broad morphologic differential ( Table 1 ). Spindle cell GISTs should be distinguished from nerve sheath tumors and smooth muscle neoplasms, whereas epithelioid GISTs may resemble malignant melanoma, paraganglioma, or carcinoid tumor. Judicious use of immunohistochemistry is key to establishing an accurate diagnosis.
| Tumor | Primary Morphology | Distinguishing Features | Immunomarkers | Other Information |
|---|---|---|---|---|
| Schwannomas | Spindle cells | Wavy fascicles; peripheral cuff of lymphocytes | Strong S-100 positivity | — |
| Desmoid fibromatosis | Spindle cells | Collagenous matrix | Nuclear β-catenin positivity in 75% | |
| Inflammatory myofibroblastic | Fascicular | Plasma cell-rich inflammatory infiltrate | ALK expression in 50% | Children and young adults |
| Smooth muscle tumors | Spindle cells | Bright eosinophilic cytoplasm; well-defined cell borders | Desmin positive | — |
| Dedifferentiated liposarcoma | Spindle cells | Pleomorphic nuclei | MDM2; CDK4 | — |
| Malignant melanoma | Variable | Intranuclear inclusions | Melan-A, HMB45, S-100 | 50% are CD117 positive |
| Angiosarcoma | Spindle cells | Highly vascular | CD31 | Commonly CD117 positive |
| Sarcomatoid carcinoma | Spindle cells | Prominent nucleoli; chromatin clearing | Cytokeratins | — |
| Carcinoid tumors | Epithelioid cells | Nested; stippled chromatin pattern | Synaptophysin, chromogranin | — |
| Paraganglioma | Epithelioid cells | Nested | Synaptophysin; S-100 | — |
| PECOMA | Epithelioid cells | — | HMB45 | — |
In 2008, a subset of GISTs was found to be immunohistochemically negative for the expression of succinate dehydrogenase subunit B (SDHB). Further studies have shown that some tumors also lack succinate dehydrogenase subunit A (SDHA) staining. The implications of these findings are discussed later.
Once a diagnosis of primary GIST has been established, the prognosis of the tumor can be assessed by taking into account 3 pathologic features: tumor size, site of origin, and mitotic index. In general, tumors arising in the stomach are less likely to recur than those arising in the small intestine or rectum. Tumors larger than 5 cm are more prone to recur or metastasize, as are tumors with more than 5 mitoses in 5 mm 2 . Tumor rupture before or during surgery is associated with a high rate of recurrence. Several risk assessment tools that incorporate these factors have been developed.
Pathology and diagnosis
GISTs most commonly arise in the stomach (60%) but also present in the small intestine (25%), rectum (5%), and other sites along the GI tract, including the esophagus, colon, appendix, and gallbladder. Rarely, the tumors appear unattached from the GI tract. These so-called extraintestinal GISTs may occupy the mesentery or omentum. GISTs are rare outside the abdominal cavity, but there is a well-documented report of a primary GIST of the pleura.
Historically, GISTs were considered to be sarcomas of smooth muscle origin, because of their predominantly spindle cell morphology and their association with the muscularis propria. However, studies using electron microscopy and immunohistochemistry suggested that these tumors differ from classic leiomyosarcoma, and in 1983, Mazur and Clark proposed the term stromal tumor. The subsequent discovery that most stromal tumors arising in the GI tract were CD34-positive provided further support for their distinction from leiomyosarcoma.
During the 1990s, several investigators noted similarities between GISTs and a little known population of cells in the gut wall designated as the interstitial cells of Cajal (ICCs). ICCs are now known to serve as pacemakers for peristaltic gut contractions. Studies during this period showed that ICCs express KIT tyrosine kinase (CD117) and are developmentally dependent on stem cell factor (SCF) signaling through this kinase. This finding led to the observation by 2 different groups in 1998 that GISTs commonly express CD117. It is now well established that 95% of GISTs are immunohistochemically positive for CD117.
In 2004, DOG-1 (also known as anoctamin 1) was described as another high-specific marker for GISTs. DOG-1 is a calcium-activated chloride channel that is highly expressed in ICCs and is detectable in 98% of GISTs, regardless of CD117 expression. Only a small percentage of other sarcomas stain positively for DOG-1. The combination of CD117 and DOG-1 expression is essentially diagnostic for GIST.
The use of CD117 and DOG-1 has helped define the range of morphologies associated with GIST. Although most GISTs are composed of a uniform population of spindled cells, some have an epithelioid appearance and others comprise a mixture of spindled and epithelioid cells. Tumor cellularity varies widely among GISTs. Low-grade lesions may show areas of central calcification, or show a bandlike alignment of nuclei that mimics a schwannoma. High-grade tumors often ulcerate the overlying mucosa and may undergo significant hemorrhagic necrosis. The variety in GIST histology dictates a broad morphologic differential ( Table 1 ). Spindle cell GISTs should be distinguished from nerve sheath tumors and smooth muscle neoplasms, whereas epithelioid GISTs may resemble malignant melanoma, paraganglioma, or carcinoid tumor. Judicious use of immunohistochemistry is key to establishing an accurate diagnosis.
| Tumor | Primary Morphology | Distinguishing Features | Immunomarkers | Other Information |
|---|---|---|---|---|
| Schwannomas | Spindle cells | Wavy fascicles; peripheral cuff of lymphocytes | Strong S-100 positivity | — |
| Desmoid fibromatosis | Spindle cells | Collagenous matrix | Nuclear β-catenin positivity in 75% | |
| Inflammatory myofibroblastic | Fascicular | Plasma cell-rich inflammatory infiltrate | ALK expression in 50% | Children and young adults |
| Smooth muscle tumors | Spindle cells | Bright eosinophilic cytoplasm; well-defined cell borders | Desmin positive | — |
| Dedifferentiated liposarcoma | Spindle cells | Pleomorphic nuclei | MDM2; CDK4 | — |
| Malignant melanoma | Variable | Intranuclear inclusions | Melan-A, HMB45, S-100 | 50% are CD117 positive |
| Angiosarcoma | Spindle cells | Highly vascular | CD31 | Commonly CD117 positive |
| Sarcomatoid carcinoma | Spindle cells | Prominent nucleoli; chromatin clearing | Cytokeratins | — |
| Carcinoid tumors | Epithelioid cells | Nested; stippled chromatin pattern | Synaptophysin, chromogranin | — |
| Paraganglioma | Epithelioid cells | Nested | Synaptophysin; S-100 | — |
| PECOMA | Epithelioid cells | — | HMB45 | — |
In 2008, a subset of GISTs was found to be immunohistochemically negative for the expression of succinate dehydrogenase subunit B (SDHB). Further studies have shown that some tumors also lack succinate dehydrogenase subunit A (SDHA) staining. The implications of these findings are discussed later.
Once a diagnosis of primary GIST has been established, the prognosis of the tumor can be assessed by taking into account 3 pathologic features: tumor size, site of origin, and mitotic index. In general, tumors arising in the stomach are less likely to recur than those arising in the small intestine or rectum. Tumors larger than 5 cm are more prone to recur or metastasize, as are tumors with more than 5 mitoses in 5 mm 2 . Tumor rupture before or during surgery is associated with a high rate of recurrence. Several risk assessment tools that incorporate these factors have been developed.
Molecular types of GIST
In 1998, Hirota and colleagues published the first report of gain-of-function mutations of KIT in GIST. KIT is the most commonly mutated oncogene in GIST, with 75% to 80% of GISTs harboring such mutations. Subsequently, it was discovered that approximately 10% of GISTs have homologous gain-of-function mutations in the PDGFRA RTK, which is a member of the same RTK family as KIT. The remaining 10% to 15% of GISTs lack mutations in either PDGFRA or KIT, and have commonly been designated as WT GIST. Recently, several other oncogenic mutations have been found in these tumors, leading us to propose that such tumors be designated as RTK-WT GIST. The molecular subtypes of GIST serve as a classification system that is useful for diagnostic, prognostic, and treatment planning purposes ( Table 2 ).
| Genetic Type | Relative Frequency (%) | Anatomic Distribution | Notable Features |
|---|---|---|---|
| KIT mutation | 77 | — | — |
| Exon 8 | Rare | Small bowel | |
| Exon 9 | 8 | Small bowel, colon | Better responses higher-dose imatinib |
| Exon 11 | 67 | All sites | Respond well to imatinib |
| Exon 13 | 1 | All sites | Imatinib responsive |
| Exon 17 | 1 | All sites | Many are imatinib sensitive |
| PDGFRA mutation | 10 | — | — |
| Exon 12 | 1 | All sites | Sensitive to imatinib |
| Exon 14 | <1 | Stomach | Sensitive to imatinib |
| Exon 18 D842V | 5 | Stomach, mesentery, omentum | Imatinib resistant |
| Exon 18 other | 1 | All sites | Some but not all are imatinib sensitive |
| RTK-WT | 13 | All sites | — |
| RTK-WT/SDHB negative | — | — | — |
| SDH mutation (A/B/C/D) | ∼2 | Stomach, small bowel | Carney-Stratakis syndrome |
| Carney triad | Rare | Stomach | Not heritable |
| Other (SDHA/B/C/D WT) | 50–70 of pediatric GIST but <2 GIST | Stomach only | Most pediatric and adults <age 30–40 y |
| RTK-WT/SDHB positive | — | — | — |
| BRAF V600E mutation | ∼2 | All sites | — |
| RAS mutations | <1 | Stomach | — |
| NF1-related | ∼1 | Small bowel | Multiple lesions, rarely malignant |
| Other | 5–10 | All sites | Most RTK-WT GIST in adults >30 y old |
KIT-mutant GIST
KIT is a type III RTK, belonging to a family that includes PDGFRA and B, CSF1R, and FLT3. Binding of dimeric SCF to the extracellular domain of KIT results in receptor homodimerization, leading to KIT tyrosine kinase activity, and subsequent activation of multiple pathways, including those involved with the PI3K/AKT and RAS signaling networks.
Normally, KIT is autoinhibited, favoring an inactive state unless bound by SCF. Mutations in the KIT receptor act to release this autoinhibition, allowing the receptor to shift into a more constitutively active state without binding SCF and thereby sustaining increased growth signaling.
The functional importance of KIT mutations in GIST pathogenesis is supported by multiple lines of evidence. First, phosphorylated KIT is almost always detectable in extracts from GIST cell lines or clinical tumor specimens. Second, mutant KIT is transforming, supporting the growth of stably transfected BA/F3 cells in nude mice. Third, when expressed in transfected cell lines, mutant forms of KIT show constitutive kinase activity in the absence of SCF, as shown by autophosphorylation and activation of downstream signaling pathways. Fourth, mice engineered to express KIT with mutations of the type found in human GISTs develop diffuse ICC hyperplasia of the stomach and intestine. These genetically modified mice also develop GIST-like tumors. This histologic picture is similar to that seen in individuals who inherit germline KIT-activating mutations. Fifth, treatment of GIST cell lines or primary GIST cell cultures with KIT kinase inhibitors or interfering RNA against KIT results in decreased proliferation and induction of apoptosis. Tyrosine kinase inhibitor (TKI)-resistant KIT-mutant GIST typically have associated secondary kinase mutations that confer drug resistance but maintain kinase activity, suggesting that even in the advanced state, such tumors require KIT kinase activation for tumor proliferation/viability (see later discussion for additional discussion of resistance mutations).
In general, GISTs are heterozygous for a given mutation; however, in approximately 15% of tumors, the remaining WT KIT allele is lost and this is associated with malignant behavior. In serial samples from individual patients, Chen and colleagues have provided evidence that this situation occurs through mitotic nondisjunction, leaving 1 daughter cell with a single chromosome 4 bearing the mutant KIT allele (uniparental monosomy).
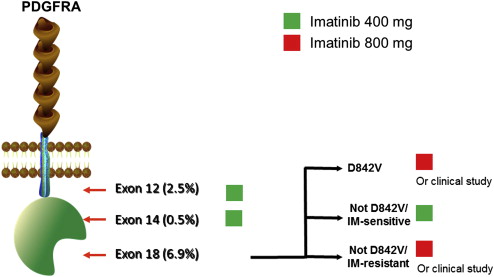
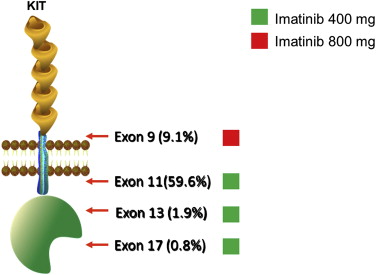
As discussed in detail later, correlative studies of outcomes during front-line treatment of metastatic disease have indicated the need to optimize therapy based on tumor genotype. Imatinib dosing recommendations for treatment of KIT-mutant GIST are summarized in Fig. 1 .
KIT exon 11 mutations
Mutations in KIT exon 11 are the most common oncogenic mutations found in GISTs, occurring in approximately 67% of cases. These mutations include in-frame deletions, insertions, or combinations thereof. Exon 11 encodes the juxtamembrane portion of KIT that normally prevents the kinase activation loop from swinging into the active conformation, thus keeping the receptor in its autoinhibited state. Mutations of KIT exon 11 disrupt this autoinhibition, allowing spontaneous kinase activation.
KIT exon 11-mutant GIST can arise anywhere in the GI tract, with the most common site being the stomach. Tumors with a KIT exon 11 mutation typically have a spindle cell appearance rather than an epithelioid morphology. After complete resection, KIT exon 11-mutant GISTs have a higher rate of recurrence than other genotypic GIST subgroups. Furthermore, GISTs with KIT exon 11 deletions have a worse prognosis than those with exon 11 point mutations, and this is particularly true for deletions involving KIT codons 557 or 558. Conversely, KIT exon 11-mutant GISTs have a more robust and durable response to treatment with the kinase inhibitor imatinib, compared with other types of GIST.
In vitro assays of KIT exon 11-mutant protein have confirmed that these mutations result in constitutive kinase activation. The KIT exon 11-mutant kinase is 10-fold more sensitive to KIT inhibitors, including imatinib, than WT KIT kinase. These in vitro findings are reflected in the clinic, where primary resistance to imatinib treatment (defined by progression within the first 6 months of therapy) is seen in only 5% of cases of advanced KIT exon 11-mutant GIST, compared with 16% in exon-9 mutant or 43% in KIT/PDGFRA WT cases. Correspondingly, the objective response rate to imatinib is 67% to 83% for KIT exon 11-mutant GIST versus 35% to 48% for KIT exon 9-mutant GIST. The median time to progression on first-line imatinib therapy for KIT exon 11-mutant GIST is approximately 25 months, and the current median overall survival for patients with KIT exon 11-mutant GIST is at least 60 months. The molecular mechanisms leading to drug resistance in KIT exon 11-mutant GISTs are discussed later.
KIT exon 9 mutations
The second most common site of mutation in GISTs is KIT exon 9 (8%–10% of GISTs), which encodes the proximal extracellular domain. More than 95% of the exon 9 mutations consist of an insertion of 6 nucleotides, resulting in duplication of amino acids 502 and 503. Rare cases of amino acid substitutions involving codon 476 have also been reported. Most GISTs harboring an exon 9 mutation are located in the small or large bowel; 25% to 30% of intestinal GISTs harbor an exon 9 mutation. In contrast, KIT exon 9 mutations are found in less than 2% of gastric GISTs.
KIT exon 9 mutations result in constitutive kinase activation and are believed to mimic the conformational change that the extracellular KIT receptor undergoes after ligand binding. The kinase domain in exon 9-mutant KIT is essentially the same as in WT KIT, and shows decreased in vitro sensitivity to imatinib compared with exon 11-mutant KIT. In agreement with the in vitro data, results from randomized phase 3 studies showed that patients with KIT exon 9-mutant GIST had a significantly improved progression-free survival when treated with a total daily dose of 800 mg of imatinib compared with patients treated with 400 mg of daily imatinib. Based on a meta-analysis of these results, the median PFS for patients with exon 9-mutant GIST was approximately 1 year longer in the 800-mg imatinib arm versus the 400-mg imatinib arm.
Other KIT mutations
Primary mutation of KIT exon 13, which encodes part of the adenosine triphosphate (ATP)-binding pocket, occurs in approximately 1% of GISTs. The substitution K642E accounts for most of these mutations. Exon 13-mutant GISTs can arise anywhere in the GI tract, but most commonly present as gastric tumors. Tumors with an exon 13 mutation typically have a spindle cell appearance, but occasionally show an epithelioid or mixed histology. The biological basis of kinase activation by this mutation has not been established, but it may interfere with normal autoinhibitory function of the juxtamembrane domain. In vitro and clinical study data indicate that this genotype is sensitive to imatinib.
Primary mutations in exon 17, which encodes the activation loop, are found in approximately 1% of GIST. Substitutions at codons 820, 822, or 823 dominate in this exon. In vitro, some of these mutations are less sensitive to imatinib than KIT exon 11-mutant kinases, but clinical responses to imatinib have been reported for primary KIT exon 17-mutant GIST. Almost all of these tumors have a spindle cell appearance, and most are located in the small bowel, but they can arise in the stomach as well.
PDGFRA-Mutant GIST
Mutations in PDGFRA are the most common non-KIT oncogenic mutations associated with GIST. PDGFRA is a tyrosine kinase receptor that is a close homologue of KIT and uses similar downstream signaling pathways. PDGFRA mutations that are found in GIST result in constitutive kinase activation. These mutations are mutually exclusive with KIT mutations. The most common locations for PDGFRA-mutant GISTs are the stomach, mesentery, and omentum, with a strong predilection for the stomach. Histologically, PDGFRA-mutant GISTs usually have an epithelioid or mixed epithelioid/spindle appearance, commonly accompanied by a myxoid stroma. PDGFRA-mutant GISTs have a lower risk of recurrence than KIT-mutant GIST. This finding explains the lower frequency of PDGFRA-mutant GIST in published series of malignant GIST versus population-based series of primary GIST. For example, a population-based series of 492 GISTs in France showed a 15% frequency of PDGFRA-mutant GIST, whereas PDGFRA-mutant GIST comprised only 2.1% of cases in 2 large clinical series of metastatic GIST. These observations have been confirmed in other series.
The biological basis for the decreased malignant potential of many PDGFRA-mutant GISTs is unclear, but these GISTs often have other characteristics of low-risk GIST, including gastric primary site, smaller size, and low mitotic index. Overall, KIT and PDGFRA-mutant GISTs show many similar features, including expression of PKC-θ, DOG-1, and activation of the RAS/MAPK and PI3K pathways. In addition, these tumors also have similar cytogenetic abnormalities, including monosomy of chromosome 14. However, gene expression profiling of KIT-mutant and PDGFRA-mutant GISTs do reveal subtle differences in the underlying tumor biology that may explain some of the differences in clinical behavior between these different entities. As with KIT mutations, rare families with germline PDGFRA mutations and susceptibility to GIST formation have been reported. Overall, the available evidence suggests that PDGFRA mutations can initiate GIST formation.
As with KIT-mutant GIST, correlative studies of outcomes during front-line treatment of metastatic disease have indicated the need to optimize therapy based on tumor genotype. Imatinib dosing recommendations for treatment of PDGFRA-mutant GIST are summarized in Fig. 2 .