For patients who survive the first 48 hours of intensive care, multiple organ failure (MOF) is the leading cause of death among patients in the intensive care unit (ICU) (see Chapter 61). Several lines of clinical evidence convincingly link gut injury and subsequent dysfunction to MOF.1 First, patients who experience persistent gut hypoperfusion after resuscitation are at high risk for abdominal compartment syndrome (ACS), which can lead to early MOF, and even death.2,3 Second, epidemiologic studies have consistently shown that the normally sterile proximal gut becomes heavily colonized with a variety of organisms. These same organisms have been identified to be pathogens that cause late nosocomial infections after the ischemia–reperfusion insult weakens the gut barriers.4,5,6,7,8,9,10,11,12 Third, gut-specific therapies (selective gut decontamination, early enteral nutrition, and most recently immune-enhancing enteral diets [IEDs]) have been shown to reduce these nosocomial infections.13,14,15,16,17,18,19 The purpose of this chapter will be to first provide a brief overview of why critically injured trauma patients develop gut dysfunction and how gut dysfunction contributes to overall morbidity and mortality. The discussion will then focus on the pathogenesis and clinical monitoring of specific gut dysfunctions. Based on this information, potential therapeutic strategies to prevent and/or treat gut dysfunction to enhance patient outcome will be discussed.
Multiple organ failure results from a dysfunctional, hyperinflammatory response producing two distinct patterns (ie, early vs late) (see Chapter 55). Soon after a traumatic insult, patients are found to be in a state of systemic hyperinflammation, now referred to as the systemic inflammatory response syndrome (SIRS).20,21,22 The intensity of SIRS is dependent upon (1) innate host factors, (2) the degree of shock, and (3) the amount of tissue injured. Of the three, shock is the predominant factor that produces a maladaptive, over exuberant SIRS response.23,24 Mild-to-moderate SIRS is most likely beneficial, whereas severe SIRS can result in early organ failure and death. As time proceeds, negative feedback systems down regulate certain aspects of acute SIRS to restore homeostasis and limit potential auto-destructive inflammation (see Chapter 61).
Reiterated in a recently published Glue Grant Study by Tompkins et al, they took blood samples of 167 patients at varying time intervals to better understand the genomic make-up after severe trauma or burn using microarray analysis. They referred to what they found as “genomic storm” in that “circulating white cells of the 167 patients studied when compared with those of the 35 normal volunteers, more than 80% of the WBC genes changed significantly.25 Tompkins et al further described the genomic patterns of complicated patients (thus that got MOF or nosocomial infections) as demonstrating higher levels of deviation from normal gene expression, and these changes quantitatively persisted longer than those same genes in the uncomplicated patients. They concluded, there were 5136 genes changed at a twofold level in the circulating WBCs of patients compared with normal volunteers. These changes were long-lasting, and in the complicated patients, more than 50% of these genes remained abnormal even at 28 days after injury.”26 The question remains what drives the inflammatory response after the acute insult to potentially cause MOF or death?
All throughout these three phenotypes of MOF (SIRS/CARS/PICS) it is hypothesized that the gut can be both an instigator and victim of the characteristic dysfunctional inflammatory response.1,6,7,9,12,18,27,28 Shock is associated with obligatory gut ischemia leading to integral damage.8 With a robust resuscitation, reperfusion results in a local intense inflammatory response that can further injure the gut, setting the stage for ACS or MOF.2,3 Additionally, the reperfused gut releases inflammatory mediators, including proteins such as cytokines and lipid such as those derived from phospholipase A2, via the mesenteric lymph, that amplify SIRS.29 Moreover, for patients undergoing laparotomy, bowel manipulation and anesthetics cause further gut dysfunction.30 Finally, standard ICU therapies (morphine, H2-antagonists, catecholamines, and broad-spectrum antibiotics) and intentional disuse (use of total parenteral nutrition rather than enteral nutrition) promote additional gut dysfunction. The end result is progressive dysfunction characterized by gastroesophageal reflux (GER), gastroparesis, duodenogastric reflux, gastric alkalinization, decreased mucosal perfusion, impaired intestinal transit, impaired absorptive capacity, increased permeability, decreased mucosal immunity, increased colonization, and gut edema. As time proceeds, the normally sterile upper gut becomes heavily colonized, mucosal permeability increases, and local mucosal immunity decreases. Intraluminal contents (eg, bacteria and their toxic products) then disseminate by aspiration or translocation to cause systemic sepsis, which then promotes further gut dysfunction.
If this dysfunction goes unchecked long enough a certain threshold is reached which propels a patient into a vicious cycle of persistent inflammatory, immunosuppressive, and catabolic state (PICS). This persistent inflammation is characterized by increased production of IL-6, a persistent acute phase response, neutrophilia with increased immature granulocyte count, anemia, lymphopenia, and, often, tachycardia.4,5 Unfortunately, this deranged inflammatory, immunosuppressive state consumes large amount of energy derived from protein catabolism. The catabolic state gives way to the new phenotype that produces substantial lean body mass loss and proportional decrease in functional status, despite aggressive nutritional support.31,32 It is the PICS phenotype that clinically depicts chronic critical illness (CCI—defined as requiring >14 days ICU care).33,34,35
A patient meets PICS criteria if he or she is residing in the ICU more than 14 days with evidence of organ dysfunction and has persistent inflammation, defined by C-reactive protein (CRP) concentration greater than 150 Hg/dL and retinol binding protein concentrations less than 1 mg/dL, immunosuppression crudely defined by a total lymphocyte count less than 0.80 × 109/L, and a catabolic state defined by serum albumin concentrations less than 3.0 g/dL, creatinine height index less than 80%, and weight loss more than 10% or body mass index less than 18 during the current hospitalization.4,5 Although these clinical markers are not direct measurements of inflammation, immunosuppression, or protein catabolism, they can serve as surrogates that are readily available in most critical care settings.
We think PICS is the new phenotype that has replaced late-appearing MOF. The major challenges for this new paradigm are: (1) to identify PICS early in its course, (2) to understand its underlying pathophysiology, and (3) to initiate appropriate multimodal therapies that target specific components of the syndrome. Medical care resource consumption associated with PICS has yet to be measured, but is likely to be a large multiple of the costs associated with the short-term treatment of trauma, severe sepsis, and septic shock.36 The incidence of PICS will likely increase, as our population ages and our ICU technology improves.37 Characterization and management of PICS will require technologies for direct monitoring and modulation of the patient’s nutritional status and immune responses. We believe that PICS is the new horizon for surgical intensive care.
To re-emphasize, after the initial insult, there is a simultaneous SIRS/CARS response. Some patients develop severe SIRS and precede to the early MOF and fulminant early death trajectory. Modern ICUs, however, are becoming increasingly effective in preventing the full expression of this phenotype (ie, early death). Some patients rapidly recover, but most patients linger in the ICU with manageable organ dysfunctions for prolonged periods, and develop CCI. A substantial portion of these CCI patients (40–60%) exhibit ongoing protein catabolism with poor nutritional status, poor wound healing, and recurrent infections.4,5 In addition, they have persistent low-grade inflammation, with defects in innate and adaptive immunity. We believe this is driven in large part by bone marrow dysfunction, with expansion and release of the myeloid derived suppressor cells (MDSCs).38
Although expansion of the MDSC population can be explained in part by increased myelopoiesis, defects in the maturation and differentiation of this cell lineage render them ineffective at fighting infections, and are responsible for producing a persistent low level inflammatory state through cytokine secretion. Through the University of Florida, Health Sepsis and Critical Illness Research Center (UF SCIRC) we will be investigating the genomic make up of PICS, trying to predict patients at high-risk of developing PICS, elucidate the role of MDSC’s, and recommend possible interventions. With a recently awarded a P50 grant by NIGMS entitled, “PICS: A New Horizon for Surgical Critical Care” the funding will provide a strong foundation to make this achievement possible. As we believe these PICS patients are ultimately discharged to LTAC facilities, rarely rehabilitate, hardly ever return to functional life, and usually experience prolonged decline and an indolent death.34,36,39,40,41,42,43,44,45,46,47,48
Intra-abdominal pressure (IAP) is monitored by urinary bladder pressure measurements. When these pressures exceed 25 cm H2O, extra-abdominal organ functions may become impaired (see Chapter 41). By definition, this is ACS. There are two types of ACS: primary (1°) and secondary (2°).2,3,49,50 Primary ACS occurs in patients with abdominal injuries that typically have undergone “damage control” laparotomy (where obvious bleeding is rapidly controlled and the abdomen is packed) and have entered the “bloody vicious cycle” of coagulopathy, acidosis, and hypothermia, which promotes ongoing bleeding (see Chapter 13). Accumulation of blood, worsening bowel edema from resuscitation, and the presence of intra-abdominal packs all contribute to increasing IAP that causes ACS. Secondary ACS occurs when extra-abdominal bleeding (eg, mangled extremity or pulmonary hilar gunshot wound) requires massive resuscitation that causes bowel edema, thus increasing IAP and eventually ACS. Markedly elevated IAP also decreases gut perfusion that may adversely affect a variety of gut functions. Clinical studies have clearly documented the poor outcome of patients developing ACS and the frequent association of ACS and MOF.2
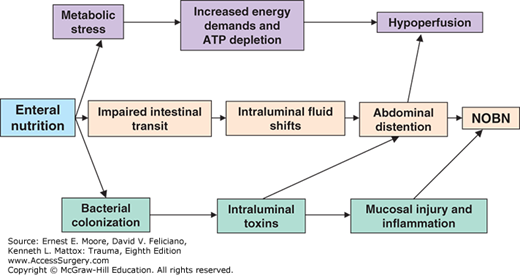
Nonocclusive small bowel necrosis (NOBN) is a relatively rare, but frequently fatal, entity that is associated with the use of enteral nutrition in critically ill patients.51 Patients typically present with complaints of cramping abdominal pain and progressive abdominal distention associated with SIRS. Computed tomography (CT) may reveal pneumatosis intestinalis or thickened dilated bowel in more advanced stages. For those who progress and require exploratory celiotomy, extensive patchy necrosis of the small bowel is found. Pathologic analysis of the resected specimens yields a spectrum of findings from acute inflammation with mucosal ulceration to transmural necrosis and multiple perforations. The consistent association with enteral nutrition indicates that inappropriate administration of nutrients into a dysfunctional gut plays a pathogenic role. There are three popular hypotheses (Fig. 58-1).51 First, metabolically compromised enterocytes become ATP depleted as a result of increased energy demands induced by the absorption of intraluminal nutrients, leading to hypoperfusion and subsequent NOBN.52 Second, hypothesis is that when nutrients are delivered into the dysmotile small bowel, fluid shifts into the lumen as a result of the presence of hyperosmolar enteral formula, leading to intestinal distention, which when severe progresses to NOBN. Third, bacterial colonization leads to intraluminal toxin accumulation, which can result in mucosal injury and inflammation, and if significant, NOBN.
The gut is a complex organ that performs a variety of functions, some of which are vital for ultimate survival of critically ill patients (eg, barrier function, immune competence, and metabolic regulation). Unfortunately, gut dysfunction in critically injured patients is poorly characterized and routine monitoring of gut function is crude. Currently, the best parameter of gut function is tolerance to enteral nutrition (see Chapter 60). For several reasons, this is an attractive parameter to monitor and potentially modulate. First, tolerance to enteral nutrition requires integrative gut functioning (eg, secretion, digestion, motility, and absorption). Second, locally administered nutrients may improve perfusion and optimize the recovery of other vital gut functions (eg, motility, barrier function, mucosal immunity). Third, tolerance correlates with patient outcome and improving tolerance will likely improve patient outcome. Fourth, refined therapeutic interventions to improve enteral nutrition tolerance will lessen the need to use parenteral nutrition and decrease its associated complications (see Chapter 60).
Gut dysfunction certainly contributors to enteral feeding intolerance. Thus, a brief overview of the pathogenesis of each of these dysfunctions and how they are monitored clinically will be reviewed to provide the rationale for proposed therapeutic strategies to improve tolerance to enteral nutrition.
Gastroesophageal reflux is an important contributing factor to aspiration of enteral feedings, which is a common cause of pneumonia in ICU patients. Reflux will occur whenever the pressure difference between the stomach and esophagus is great enough to overcome the resistance offered by the lower esophageal sphincter. Increases in gastric pressure can be due to distention with fluids and failure of the stomach to relax to accommodate fluid. Decreases in resistance at the lower esophageal sphincter can be due to relaxation of the sphincter muscle in response to many stimuli including mediators released during injury and resuscitation. Additional contributing factors include (1) forced supine position, (2) the presence of a nasoenteric tube, (3) hyperglycemia, and (4) morphine.
Commonly used clinical monitors include laboratory testing for presence of glucose in tracheal secretions or by observing blue food dye, which has been added to the enteral formula in tracheal aspirates. Detection of glucose lacks specificity. False-positive results can occur with high serum glucose levels or presence of blood in tracheal secretions. The use of blue food dye is poorly standardized and lacks sensitivity. More importantly, however, several reports document absorption of blue food dye in critically ill patients that is associated with death. This is presumably due to a toxic effect that blue food dye has on mitochondrial function. A recent consensus conference recommended that both these techniques be abandoned.53 Unfortunately, there are no simple monitors of GER other than observing for vomiting or regurgitation, which are not very sensitive. The head of the bed should be elevated 30° to 45° to decrease the risk that when GER occurs that it is less likely to result in pulmonary aspiration (see Chapter 57). Gastric residual volumes (GRVs) should be monitored with the presumption that a distended stomach will lead to higher volume GER (see Chapter 66).
Gastroparesis is common in ICU patients and predisposes to increased duodenogastric reflux (a potential contributing factor for gastric alkalinization) and GER (a contributing factor for aspiration). The mechanisms responsible for gastroparesis in critical illness have not been well studied. Potential factors include (1) medications (eg, morphine, dopamine), (2) sepsis mediators (eg, nitric oxide), (3) hyperglycemia, and (4) increased intracranial pressure.
The common clinical monitors for gastroparesis are intermittent measurement of GRVs when feeding into the stomach or measurement of continuous suction nasogastric tube output when feeding postpyloric. The practice of using GRV is poorly standardized and is a major obstacle to advancing the rate of enteral nutrition.54 GRVs appear to correlate poorly with gastric function and GRVs less than 200 cc generally are well tolerated. GRVs of 200–500 cc should prompt careful clinical assessment and the initiation of a prokinetic agent. With GRVs more than 500 cc, enteral nutrition traditionally was stopped. After clinical assessment excludes small bowel ileus or obstruction, placement of a postligament of Treitz feeding tube should be considered (see Chapter 60). However, a study by Reignier et al, in JAMA should that not monitoring GVR and allowing patients to advance to goal tube feed rates for adequate nutritional supplementation was not inferior to checking GVR in preventing ventilator associated pneumonia.55
Although not well studied in trauma specifically, critically ill patients are known to have a high incidence of gastroduodenal reflux. In a study of antral, duodenal, and proximal jejunal motility, Tournadre et al demonstrated that postoperative gastroparesis occurs after major abdominal surgery and is associated with discoordinated duodenal contractions of which 20% migrated in a retrograde fashion.56 Heyland et al administered radio-labeled enteral formulas through a standard postpyloric nasoenteric feeding tube in ventilated ICU patients and documented an 80% rate of radio isotope label reflux into the stomach, 25% reflux rate into the esophagus, and a 4% reflux rate into the lung.57 Finally, Wilmer et al reported monitoring bile reflux in the esophagus of ventilated ICU patients using a fiberoptic spectrophotometer that detects and quantifies bilirubin concentration.58 Endoscopy was performed and documented erosive esophagitis in half of the patients of which 15% had pathologic acid reflux and 100% had pathologic bile reflux. These studies provide convincing evidence that duodenogastric reflux is a common event in ICU patients.
The stomach, through secretion of hydrochloric acid, normally has a pH below 4.0. This acid environment has been correlated with the relatively low bacterial counts found in the stomach. Reviews of several studies have shown that alkalinization of the stomach through the use of antacids, H2 antagonists, and proton pump inhibitors results in gastric colonization by bacteria not normally found in the stomach; and several, but not all, studies have shown that gastric colonization predispose patients to ventilator-associated pneumonia,59 and can increase the risk of community-acquired Clostridium difficile–associated disease.60
Several animal studies conducted by our group have shown that both lipopolysaccharide administration and mesenteric ischemia/reperfusion result in the gastric accumulation of an alkaline fluid.61 This most likely results from a decrease in gastric acid secretion with continued gastric bicarbonate secretion and the reflux of duodenal contents into the stomach. Even more recently, we have reported that the pH of gastric contents in trauma patients also is elevated, possibly due to similar events.62 Thus, even without the administration of antacids or inhibitors of acid secretion, gastric alkalinization and bacterial colonization of the stomach are likely in this group of patients. When this is combined with the gastroparesis often seen in these patients (see above), it is easy to envision the stomach becoming a major source of bacteria for ventilator-assisted pneumonia and perhaps translocation to other organs.
Shock results in disproportionate splanchnic vasoconstriction. The gut mucosa appears to be especially vulnerable to injury during hypoperfusion. The arterioles and venules in the small bowel mucosal villi form “hairpin loops.” This anatomic arrangement improves absorptive function, but it also permits a countercurrent exchange of oxygen from the arterioles to the venules in the proximal villus. Under hypoperfused conditions, a proximal “steal” of oxygen is believed to reduce the Po2 at the tip of the villi to zero. The gut mucosa is further injured during reperfusion by reactive oxygen metabolites and recruitment of activated neutrophils (see Chapter 61). This mucosal injury, however, appears to quickly repair. Mucosal blood flow does not always, though, return to baseline with resuscitation and this is in part due to defective vasorelaxation. The gut mucosa is also vulnerable to recurrent episodes of hypoperfusion from ACS, sepsis, and use of vasoactive drugs. Whether recurrent hypoperfusion results in additional ischemia/reperfusion injury is not known, but it is reasonable to assume that hypoperfusion would decrease gut nutrient absorption and render the patient more susceptible to NOBN.
Monitoring gastric mucosal perfusion in the clinical setting can be done by gastric tonometry. With hypoperfusion, intramucosal CO2 increases due to insufficient clearance of CO2 produced by aerobic metabolism or due to buffering of extra hydrogen ions produced in anaerobic metabolism. As intramucosal CO2 accumulates, it diffuses into the lumen of the stomach. The tonometer measures the CO2 that equilibrates in a saline filled balloon (newer monitor uses air-filled balloon) that sits in the stomach. This is the regional CO2 tension (PrCO2) and is assumed to equal the intramucosal CO2 tension. Using this measured PrCO2 and assuming that arterial bicarbonate equals intramucosal bicarbonate, the intramucosal pH (pHi) is calculated by using the Henderson–Hasselbalch equation. Numerous studies have documented that a persistently low pHi (or high PrCO2 level) despite effective systemic resuscitation predicts adverse outcomes and that attempts to resuscitate to correct a low pHi do not favorably influence mortality.63 Unfortunately, alternative resuscitation strategies have not been able to increase pHi to improve outcome and thus this monitoring tool is in search of a novel application (see Chapter 55).
Laboratory models of shock, bowel manipulation, and sepsis demonstrate that small bowel transit is impaired.64 This impairment in turn is expressed as a decrease in the number and/or force of contractions, or as an abnormal pattern of contractions. Although the results in animal models are convincing, surprisingly, clinical studies indicate that small bowel motility and transit are more often than not well preserved after major elective and emergency laparotomies.65 This observation coupled with the observation that small bowel absorption of simple nutrients is relatively intact provided the rationale for early jejunal feeding (see Chapter 60).
Clinical studies have documented that the majority of critically ill patients tolerate early jejunal feeding.66 In a recent study, severely injured patients had jejunal manometers and feeding tubes placed at secondary laparotomy.65 Surprisingly, 50% had fasting patterns of motility that included components of the normal migrating motor complexes (MMCs). These patients tolerated advancements of enteral nutrition without problems. The other 50% who did not have fasting MMCs did not tolerate early advancement of enteral nutrition. Of note, none of the patients converted to a normal-fed pattern of motility once they reached full-dose enteral feeding. This could be due to infusion of caloric loads insufficient to bring about conversion. On the other hand, the failure to develop fed activity, a pattern of motility promoting mixing and absorption, might explain why diarrhea is a common problem in this patient group.
Although manometry can be used to monitor motility, it is not practical. Unfortunately, simpler indicators of motility such as the presence of bowel sounds or the passing of flatus are unreliable. Other minimally invasive methods to monitor transit are needed. Contrast studies through the feeding tubes are relatively simple, but not validated.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


