Fig. 12.1
Age-standardized colorectal cancer incidence rates by World Health Organization region and sex for 2008 [4]
Mortality rates in the United States have declined since the mid-1980s, due to a combination of early detection and better treatment options [5]. The United States has one of the highest survival rates from colorectal cancer in the world with a 5-year survival rate of 61 % for all patients treated, including all stages and sites combined [6]. Outside the United States, mortality rates continue to climb, however, as the incidence increases without significant improvement in screening or therapeutic options [7].
Gastric cancer remains one of the most common cancers worldwide, although its incidence is now decreasing worldwide [1]. It was the leading cause of cancer deaths in the world until the increase incidence and mortality of lung cancer in the 1980s [8]. It still remains the fourth leading cause of cancer-related deaths, accounting for an estimated 10 % of all cancer deaths worldwide [1]. Gastric cancer has several recognized risk factors including H. pylori and dietary and environmental factors and the decline in its incidence can be attributed in part to elimination of people’s exposure to these factors [9]. While the overall incidence is declining, the absolute number of new cases annually is increasing, most likely due to world’s aging population. Additionally, the incidence is actually increasing, by almost 60 %, in younger patients ages 25–39 years [10]. Thus, gastric cancer continues to have a great impact, both globally and in the United States.
While the vast majority of gastric and colorectal cancers are adenocarcinomas, esophageal cancers are primarily either squamous cell or adenocarcinomas. The incidence of squamous cell carcinoma of the esophagus is decreasing in the United States, but the incidence of adenocarcinoma is increasing [11]. Incidence of esophageal cancer varies by geographically, with the highest rates found in Southern and Eastern Africa and Eastern Asia and the lowest rates in Western and Middle Africa and Central America [1]. In the highest-risk areas, the major histology is squamous cell carcinoma [12]. In Western countries, the incidence of adenocarcinoma is increasing, in part due to the increase incidence of gastroesophageal reflux disease and obesity, known risk factors [13, 14].
As better understanding of the molecular changes specific to carcinogenesis have been derived, the concept of targeted therapy has arisen in oncology. With better understanding, new treatments have been developed. Perhaps the most striking example is the significant in overall survival in patients with metastatic colorectal cancer with the introduction of targeted therapy. Median survival for patients with metastatic colorectal cancer now ranges from 24 to 28 months, which is an approximate fourfold increase over the last 20 years [15] (Fig 12.2).
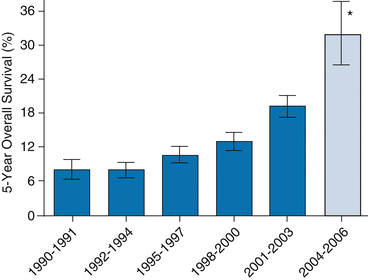
Fig. 12.2
Median overall survival for patients with metastatic colorectal cancer treated at the M.D. Anderson Cancer Center and the Mayo Clinic by year of diagnosis (error bars are 95 % confidence intervals) [16]
Additionally, molecular testing allows oncologists to predict patients who will not respond to certain therapies, for example the identification of KRAS mutation and the use of EGFR-targeted monoclonal antibodies, as discussed in more detail below. However, as these therapies are effective only if the particular mutation(s) are present, it is of the utmost importance that the proper results of gene or protein assays are obtained. There is immense clinical relevance and without an accurate molecular analysis by the pathologist, there may be opportunities lost. These opportunities can take the form of using a targeted agent that is expected to give a positive response in diseases with a specific molecular milieu. Alternatively, and just as important, if the specific genetic aberrations required for a therapy to be effective are not present, identification of this may save a patient from side effects they would otherwise endure in order to gain some benefit. We will discuss the major assays for GI oncology.
ERBB2 (HER2)
The human epidermal growth factor receptors (HER) are transmembrane proteins with an extracellular domain that may bind ligands and an intracellular tyrosine kinase domain [17]. Ligand binding induces conformational changes and dimerization, which activates the kinase activity intracellularly and the subsequent downstream signaling cascade. The extracellular domain of ERBB2 (also known as HER2), one member of the four HER family proteins, remains in an perpetually active conformation, allowing ERBB2 (HER2) to dimerize with other HER family proteins [18]. It is known that overexpression of ERBB2 (HER2) in breast cancer leads to increased dimerization, producing a strong pro-tumorigenic signaling cascade [19].
Role of Molecular Testing
For patients with inoperable locally advanced, recurrent, or metastatic adenocarcinoma of the esophagus, GE (gastroesophageal) junction and stomach for whom trastuzumab therapy is being considered, assessment for tumor ERBB2 (HER2) overexpression using immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH) or other in situ hybridization methods is recommended [20].
While the prognostic role of ERBB2 (HER2) is well established in breast cancer, it is less clear in gastric cancer [21]. The level of ERBB2 (HER2) gene amplification in advanced gastric cancer correlates with predicted response to trastuzumab-based therapy [22].
The first targeted agent with documented efficacy in advanced gastric and gastroesophageal junction cancer was trastuzumab, the humanized monoclonal antibody against ERBB2 (HER2) [23]. Based on the preclinical observations that approximately 20 % of gastric cancers (and approximately 30 % of gastroesophageal adenocarcinomas) over express ERBB2 (HER2) the phase III study of trastuzumab in Gastric Cancer (ToGA) investigated whether the addition of trastuzumab to standard chemotherapy would extend survival in patients with advanced adenocarcinoma of the stomach or gastroesophageal junction [24, 25]. The ToGA trial was the first phase III trial to demonstrate a survival advantage with the addition of a biologic agent, trastuzumab, to standard chemotherapy in advanced gastric cancer. Further phase III trials are investigating the role of other EGFR/ERBB2 inhibitors (Fig. 12.3).
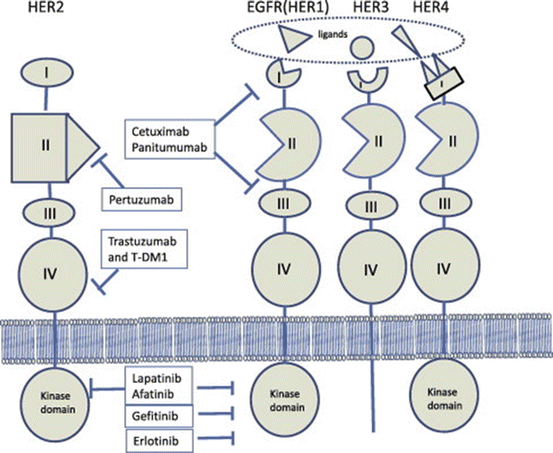
Fig. 12.3
HER family receptors and anti-HER (ERBB2) agents approved in clinical practice. Anti-HER agents include: (1) Pertuzumab, which is specific to HER2, binding to it and blocks its dimerization. (2) Cetuximab. (3) Panitumumab, both which bind to EGFR and block its ligand binding. (4) Trastuzumab. (5) Ado-trastuzumab emtansine (T-DM1) both which block HER2 signaling by binding to the juxtamembrane domain of HER2. T-DM1 also releases the cytotoxic anti-microtubule moiety (DM1) after internalization of HER2:T-DM1 complex. (6) Gefitinib. (7) Erlotinib both which are small molecular tyrosine kinase inihibitors which inhibit the cytoplasmic kinase domain in EGFR. (8) Lapatinib. (9) Afatinib target kinase domain of both EGFR and HER2 [26]
Methodology
Testing of all patients with advanced adenocarcinoma of the esophagus, GE junction, and stomach, for ERBB2 (HER2) overexpression should be considered. Testing can be performed by fluorescence in situ hybridization (FISH) or immunohistochemistry. Immunohistochemistry testing is the primary method used in hospitals [27]. Immunohistochemistry should be the initial test and fluorescence in situ hybridization should be used to retest IHC 2+ samples [28].
Criteria for ERBB2 (HER2) status are dependent on the tumor type (Table 12.1). For example, criteria differ between breast and gastric cancer due to tumor heterogeneity and membrane staining in gastric cancer [24]. In the US, trastuzumab added to chemotherapy is FDA approved for metastatic gastric cancer if FISH positive or 3+ IHC and 2+ ICH/FISH+. This utilizes the criteria of the ToGA trial. See Fig. 12.4.
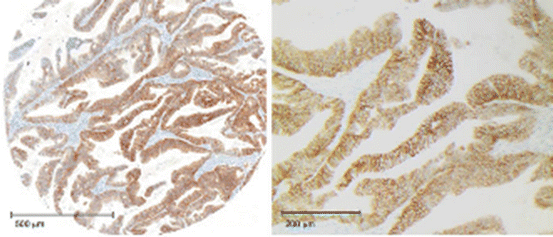
Table 12.1
Immunohistochemical criteria for scoring ERBB2 (HER2) expression in gastric and esophogastric junction cancers [29]
Surgical specimen expression pattern, immunohistochemistry | Biopsy specimen expression pattern, immunohistochemistry | ERBB2 (HER2) overexpression assessment | |
|---|---|---|---|
0 | No reactivity or membranous reactivity in <10 % of cancer cells | No reactivity or no membranous reactivity in any cancer cell | Negative |
1+ | Faint or barely perceptible membranous reactivity in ≥10 % of cancer cells; cells are reactive only in part of their membrane | Cancer cell cluster with a faint or barely perceptible membranous reactivity irrespective of percentage of cancer cells positive | Negative |
2+ | Weak to moderate complete, basolateral, or lateral membranous reactivity in ≥10 % of cancer cells | Cancer cell cluster with a weak to moderate complete, basolateral, or lateral membranous reactivity irrespective of percentage of cancer cells positive | Equivocal |
3+ | Strong complete, basolateralor lateral membranous reactivity in ≥10 % of cancer cells | Cluster of five or more cancer cells with a strong complete, basolateral, or lateral membranous reactivity irrespective of percentage of cancer cells positive | Positive |
Fig. 12.4
IHC score 3+. Left: visible by naked eye, with membranous staining clearly visible at low magnification (obj. ×5). Right: Staining either complete, basolateral, or lateral (×10) [29]
Both surgical specimens and biopsy samples are acceptable for ERBB2 (HER2) testing. A sufficient amount of sample should be tested due to heterogeneity of expression of ERBB2 (HER2) in gastric cancer, in order to avoid false negative results [28]. Samples for HER2 testing should be fixed immediately, as time from biopsy or excision to placement in formalin fixative can affect HER2 testing results. Additionally, poor fixation is often an important source of error in HER2 testing [30]. Both positive and negative controls should be performed whenever immunohistochemistry is done [28]. Performing testing in a centralized reference laboratory or a laboratory with reasonable volume and expertise is important for quality assurance and to obtain quality HER2 testing results [28] (Figs. 12.5 and 12.6).
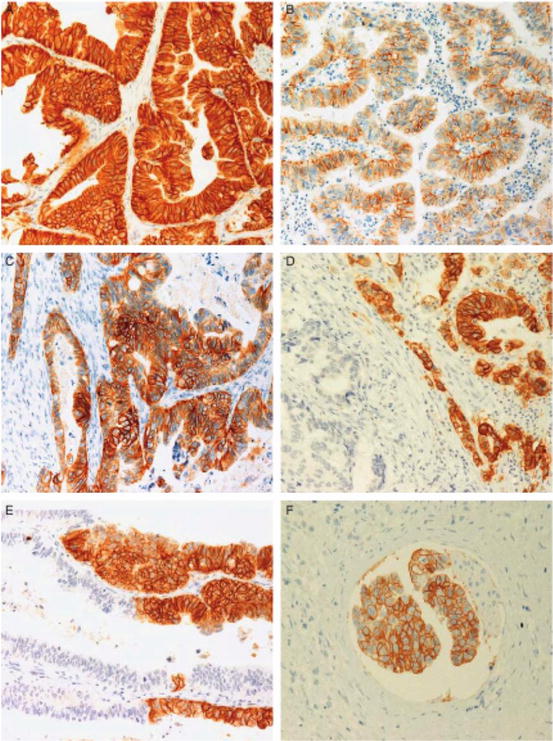
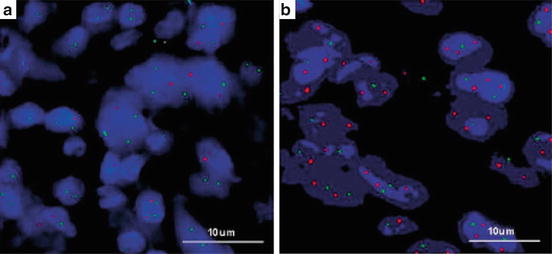
Fig. 12.5
Examples of ERBB2 (HER2) IHC expression in gastric carcinoma. (a) 3+ HER2 reactivity with intense membrane staining, but no luminal reactivity. (b) 2+ HER2 reactivity with moderate intensity, mainly lateral portions of membrane. (c) HER2 staining from 3+ to 1+. (d) HER2 3+ component adjacent to a negative area, showing different morphologic aspects. (e) HER2 3+ neoplastic cells intermixed with negative cells. (f) HER2 3+ neoplastic embolus with intestinal-type features surrounded by HER2- diffuse-type adenocarcinoma [31]
Fig. 12.6
(a, b) ERBB2 (HER2) gene amplification detected by FISH. A ratio of ERBB2 (HER2) (red): CEP17 (green) <2.0 was considered nonamplified and ratios >/=2.0 were considered amplified [32]
Interpretation of Results
Recommendations for interpreting ERBB2 (HER2) IHC in gastric specimens are available. One uses ability to visualize membrane staining at certain magnifications as a guideline for scoring [29]. For example, if membrane staining can be seen with the naked eye or 5× microscopic magnification, this usually correlates with a 3+ score. If a higher magnification is needed, 20× or 40×, staining is 1+ or negative.
Evaluation of ERBB2 (HER2) status in gastric cancer is different than in breast cancer. In gastric cancer there is infrequent completeness of membrane staining, whereas it is required in breast cancer [31] (Fig. 12.7).
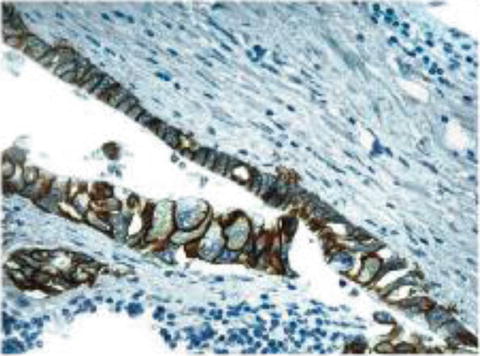
Fig. 12.7
Incomplete membrane staining may still be ERBB2 (HER2) positive in gastric cancer, whereas it represents HER2 negative disease in breast cancer [24]
Also incongruent from breast cancer, there is heterogeneity of immunostaining in gastric adenocarcinoma. If immunohistochemical testing is borderline or there is felt to be a problem with the sample, retesting via fluorescence in situ hybridization should be done. Additionally, if there is strong HER2 staining, but in less than 10 % of cells, also consider repeating with FISH [28] (Fig. 12.8).
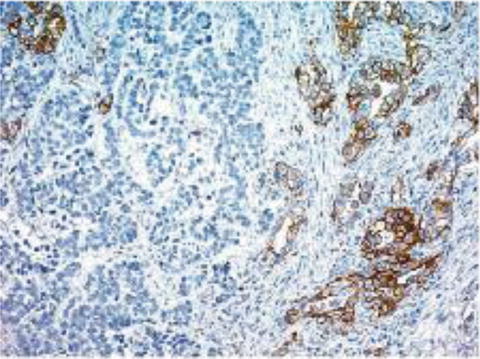
Fig. 12.8
IHC staining for ERBB2 (HER2), illustrating tumor heterogeneity. This can be a cause for discrepancies between IHC and FISH assays, where IHC may be read as IHC 0, but FISH would be positive [24]
Microsatellite Instability (MSI)
Microsatellite instability (MSI) is an important molecular concept in colon cancer. Microsatellites are sections of DNA where a short sequence of nucleotides are repeated many times [33]. MSI results from the loss of mismatch repair protein (MMR) expression. Without mismatch repair proteins, the ability to consistently replicate repetitive DNA sequences is lost. This results in variations in the lengths of microsatellite DNA sequences in tumor cells. These tumors are termed MSI-H-instability phenotype, describing their high level of microsatellite instability. Germline mutations in any of the genes that enable mismatch repair result in the development of hereditary nonpolyposis colorectal cancer (HNPCC). This is an autosomal dominant disorder that confers an 80 % lifetime risk of the development of colorectal cancer [34]. These tumors tend to be found on the right side of the colon and be high-grade, mucinous type [35]. They characteristically have increased lymphocyte infiltration [36].
Role of Molecular Testing
MSI-H phenotype is a favorable prognostic indicator for 5-year disease-free survival [37]. Patients with MSI-H colorectal cancers have a better long-term prognosis than patients who have the same stage disease, but microsatellite stability [38].
Microsatellite instability is an important prognostic indicator in patients with Stage II colon cancer, as adjuvant chemotherapy is beneficial to only a small portion of these patients. It is a contributing factor in 15 % of all sporadic colon cancers [39]. Patients with tumors with microsatellite instability have been found to not benefit from adjuvant 5-fluoruracil-based chemotherapy [40]. Another study also showed that patients with preserved mismatch repair had an overall survival benefit when treated with adjuvant 5-fluorouracil chemotherapy. Conversely, patients with deficient MMR had a worse overall survival when treated with 5-fluorouracil versus observation alone [41].
Methodology
Tumors with mismatch repair protein deficiencies can be identified by either immunohistochemistry for MMR proteins or polymerase chain reaction assessment of DNA markers for microsatellite instability [42]. Formalin-fixed, paraffin-embedded tissues sections can be used with mouse primary antibodies using standard immunohistochemical techniques [43] (Fig. 12.9).
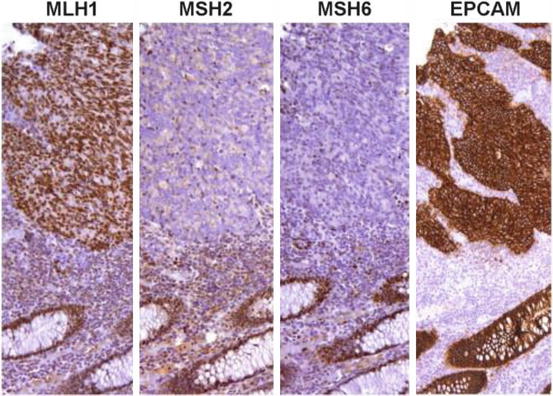
Fig. 12.9
Medullary carcinoma with LS-R immunohistochemical pattern: MLH1+, MSH2−, MSH6−, and EPCAM+ [43]
MSI analysis requires isolation of tumor DNA. PCR can then be performed using known microsatellite primers (NR21, NR24, NR27, BAT25, and BAT26) [44]. The Bethesda guidelines were developed to isolate patients who may have HNPCC based on their clinical presentation and family history. There is a panel of mononucleotide and dinucleotide repeats that was considered the “reference panel” after the 1997 National Cancer Institute (NCI) workshop produced these recommendations (BAT25, BAT 26, D2SI23, D5S346, and D17S250) [45]. New panels have been developed since then, some of which have higher sensitivity and positive predictive value than the NCI panel [46]. After limitations were seen with the Bethesda panel, a new pane of five quasi-monomorphic mononucleotide markers was developed (BAT-26, BAT-25, NR-21, NR-22, NR-24) [47]. This is known as the pentaplex panel and found to have increased sensitivity and specificity compared to the Bethesda panel [48]. It is the current panel recommended for MSI testing after another NCI conference was held in 2002, although new panels continue to be developed. For example, there has now been a hexaplex panel developed using coamplification of six mononucleotide repeat markers (BAT25, BAT26, BAT40, NR21, NR22, NR27) and one polymorphic dinucleotide marker (D3S1260) [49] (Fig. 12.10).
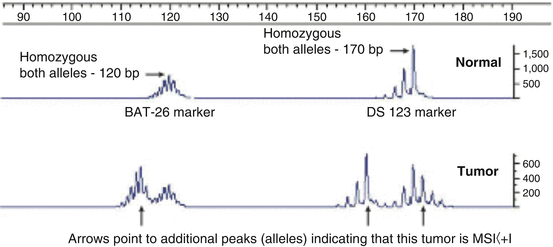
Fig. 12.10
Microsatellite instability determination of two microsatellites in the same multiplexed run. The upper tracing is from blood DNA showing BAT26 at 120 bp and DS123 at 170 bp. The patient is homozygous for both markers. The lower tracing is from the same patient’s tumor DNA [50]
Testing should be considered in all patients less than 50 years old or with Stage II disease [41]. Younger patients should be tested as their disease may be the result of HNPCC. Patients should be tested for MSI if they have Stage II disease as it provides important information about the role of adjuvant chemotherapy.
Epidermal Growth Factor Receptor (EGFR)
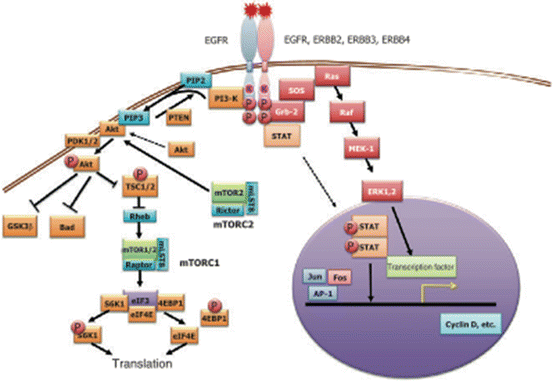
The epidermal growth factor receptor (EGFR), also called HER1, is a transmembrane receptor. When a ligand binds, the receptor dimerizes, stimulating the phosphorylation of the tyrosine kinase on the intracellular domain [51]. This initiates a signaling cascade, which is responsible for cell proliferation, migration, adhesion, differentiation, and survival [52]. The binding of ligands to EGFR initiates cell signaling through several different pathways, including the Ras-Raf-mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K)-Akt, and phospholipase C pathways [17, 53]. There are currently two monoclonal antibodies available that target EGFR, cetuximab, and panitumumab (Fig. 12.11).
Methodology
Fluorescence in situ hybridization (FISH) assay has been performed to determine the number of gene copies of EGFR. There was a positive correlation with increasing number of EGFR gene copies having an increased response to cetuximab [55]. However FISH testing is limited by the lack of a clear cutoff point for interpretation. This makes its implementation in clinical practice very limited. Immunohistochemistry analysis has also been investigated, but studies now show there is no predictive value of EGFR testing in colorectal cancer to determine response to EGFR inhibitors [56]. Multiple studies have shown that EGFR expression, as determined by immunohistochemistry does not correlate clinical response to EGFR inhibitors [57]. Technical reasons have been identified as a reason for the lack of association between EGFR detection by immunohistochemistry and response to EGFR inhibitors [58]. Some reasons include the EGFR detected by immunohistochemistry may not be the same targeted by anti-EGFR treatments. Processing and storage of the tumor sample may also affect immunohistochemistry results, producing a discrepant EGFR result from the patient’s outcome perhaps. As such, there is no clinical role for EGFR immunohistochemistry testing. Without clinical indication, it is not routinely performed.
Another analysis option is FISH, which can detect the small proportion of colorectal tumors which have an overexpression of EGFR [59]. The number of gene copies of EGFR per nucleus seems to have a direct correlation with response to EGFR inhibitors [60]. While there is suggestion of clinical relevance in EGFR detection, testing by either immunohistochemistry or FISH is not standardized yet and should not be routinely performed to assist with clinical decision making.
KRAS
Kirsten rat sarcoma viral oncogene homolog (KRAS) is an oncogene. It is a part of the Ras family which globally produces growth factors. They are activated in a large variety of cancers. KRAS gene mutations are found in about 40 % of colorectal carcinomas [61]. KRAS mutations have effects on signaling pathways for cell proliferation, survival, and metastasis. KRAS can cause a growth factor signaling cascade through the mitogen-activated protein kinase (MAPK) pathway that controls the G1-S cell cycle transition [62]. KRAS also signals through the phosphatidylinositol 3-kinase (PI3K) pathway, which will be discussed later [63]. KRAS is a signal transduction protein that serves as in-between signals from EGFR to the nucleus. If KRAS is mutated, the signal pathway is always turned on, regardless of inhibitor of EGFR. Therefore, EGFR inhibitors, like cetuximab and panitumumab are ineffective in patients with KRAS mutations. This observation is supported by several clinical trials [64, 65]. KRAS mutations are point mutations found at codons 12 and 13 with the majority of mutations found at codon 12 [66] (Fig. 12.12).
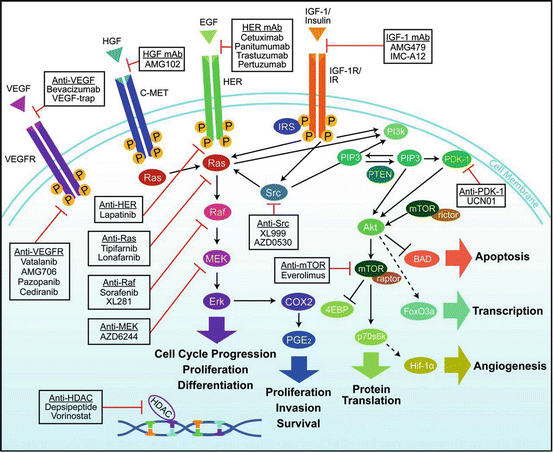
Fig. 12.12
Overview of cellular signaling pathways involved in the proliferation and progression of colorectal cancer. Agents targeting signaling proteins that have been evaluated or are currently being evaluated in phase II, III, or IV clinical trials for colorectal cancer are shown. The epidermal growth factor receptor (EGFR)-related family of receptor tyrosine kinases includes EGFR (ERBB1); ERBB2 (HER2); ERBB3 (HER3); ERBB4 (HER4). MET (C-MET) MET proto-oncogene, receptor tyrosine kinase; EGF epidermal growth factor; HDAC histone deacetylases; HGF hepatocyte growth factor; IGF1 insulin-like growth factor 1 (somatomedin C); IGF1R insulin-like growth factor 1 receptor; VEGFA (VEGF) vascular endothelial growth factor A; KDR (VEGFR) kinase insert domain receptor [57]
Because of EGFR inhibitors are only effective in KRAS wild-type tumors, KRAS mutation status determination is critical in determining treatment options in advanced colorectal cancer. KRAS mutations have no role in adjuvant therapy and therefore are not required unless patient has metastatic disease. Genotyping for KRAS mutation in all patients with stage IV, metastatic disease is standard practice [67]. KRAS mutation status determination prior to using cetuximab or panitumumab is recommended by both the US Food and Drug Administration and American Society of Oncology [68, 69, 67]. The use of EGFR inhibitors in KRAS mutated disease has been associated with worse outcomes, perhaps due to increased toxicities without any benefit to the disease [70, 56]. Again, this illustrates the need for upfront KRAS mutation testing in metastatic colorectal cancer. There is currently no role in obtaining KRAS mutation in Stage I, II, or III colorectal cancer, as EGFR inhibitors are not standard of care in these settings.
KRAS mutation analysis has clinical relevance because of the ability to use EGFR inhibitors in the disease. One study, looking at cetuximab versus best supportive care in metastatic chemotherapy-refractory colorectal cancer, overall survival in patients with KRAS wild-type was 9.5 months with cetuximab versus 4.8 months with best supportive care alone. This is in contrast to patients with KRAS mutation, where overall survival was 4.5 months with cetuximab and 4.6 months with best supportive care [65].
The prognostic value of KRAS mutations is not clearly established. Studies have shown that KRAS mutations generally confer a worse prognosis, while later analysis showed it was the glycine to valine substitution at codon 12 that was specifically associated with poorer prognosis. Subsequent studies have had mixed results on association of KRAS mutation status and patient outcome. It is still felt that KRAS-mutated colorectal cancers may have a poorer prognosis overall [71]. As the absence of KRAS mutation predicts response to anti-EGFR therapy, it does hold prognostic value that way, as response to these therapies improves survival [72].
While it is known that most KRAS mutations will be found at codons 12 and 13, other mutations have been found, including A146 mutations and codon 61. These mutations result in a similar resistance to EGFR inhibitor therapy. Identification of these less common mutations would help to more comprehensively guide therapy.
Methodology
Testing for KRAS should be performed only in laboratories qualified to perform high-complexity clinical laboratory (molecular pathology) testing, as certified under the clinical laboratory-specific amendments of 1988 (CLIA-88). The therascreen KRAS RGQ PCR Kit (Qiagen) is the only FDA-approved test for KRAS mutational analysis. It detects seven common mutations within codons 12 and 13 [73]. Amplification refractory mutations system (ARMS) and Scorpions primers identify the mutations [74].
Most assays for KRAS mutations allow testing on formalin-fixed paraffin-embedded tissue. Testing can be performed on either primary colorectal cancers or metastases [75]. Nuclei-rich samples with a low level of necrotic tissue, often the deepest invasive portion of the tumor for sampling may help increase sensitivity of testing [73]. The pathologist should chose samples that consist of mostly tumor cells with as little necrosis or inflammation as possible. Three types of samples can be obtained: (1) freshly extracted from patient, provided fresh or in RNA preservative, (2) freshly extracted from the patient and rapidly frozen and (3) neutral buffered formalin fixed and paraffin embedded [67]. The latter is the most clinically relevant as that is how such specimens are handled after they have been obtained.
There is no specific methodology recommended. Real-time polymerase chain reactions, direct sequencing analysis, or hybridization can be used to determine KRAS mutation status [76]. There are several kits available [67]. It is expected that companies will seek US Food and Drug Administration approval for commercially available assays. See Table 12.2.
Method | Technology | Sensitivity, MT/WT%a | Time to result | Pros | Cons |
|---|---|---|---|---|---|
Direct sequencing | |||||
Cycle sequencing | Singer sequencing using dye-labeled dideoxynucleotide chain termination | 15–25 | 4 days to 2 weeks (paraffin) | Gold standard Detects all mutations | Insensitive Labor intensive |
Pyrosequencing | Measures pyrophosphate release during DNA extension | 5–10 | Fast | High-throughput Precise/reproducible Suitable for partially degraded samples | Expensive |
PCR-based methods | |||||
ARMSa | Mutation-specific PCR amplification | 1 | Rapid: <2 days (paraffin) | High sensitivity. Rapid results | Detects single mutation per reaction Requires engineered primer/probe |
TheraScreenb | Combination of ARMS, Scorpionab (allele-specific probel, and real-time PCR) | 1–5 | Rapid: 2 days 2 h to process samples | Rapid results High sensitivity Commercially available | Detects only seven common mutations Requires more tissue Very expensive |
Allele-specific oligonucleotide hybridization | |||||
Allele-specific probes | Probes hybridize to wild-type or mutant sequence impacting melting temperature | 10 | Rapid: <2 days (paraffin) | Rapid results | Low sensitivity |
ViennaLabb | Hybridization of PCR products to array of allele-specific oligonucleotides | 1 | Rapid: 6 h to process samples | Detects 13 common mutations Less expensive than TheraScreenb | Complicated data interpretation |
The International Organization for Standardization and the College of American Pathologists have suggested specific information that should be recorded on each report, including: the patient, the specimen, the ordering physician, and the laboratory where testing was done. Completeness is essential as it can otherwise commonly lead to inaccurate interpretation of KRAS testing [78]. The specific mutation that is observed should be noted, because it may have an impact on therapy, especially in the future. New mutations are being found, the significance of which is not yet known. Additionally, some mutations may not confer as much resistance to EGFR inhibitors as others [79].
The American Society of Clinical Oncology recommends that the KRAS assay be reported as KRAS normal, meaning no mutation was identified and KRAS abnormal, meaning a mutation was found and treatment with anti-EGFR monoclonal antibody therapy is not recommended. The report should specify which mutation was found. All reports should specify what assay was done and what controls were used [67]. As the field continues to evolve and more clinical information is gained about the prognostic and therapeutic implications of molecular testing, it is important to communicate to the oncologist exactly which KRAS mutation is present.
Insufficient quantity and/or poor quality of the DNA template are the most likely reasons for test failures of RAS mutations [80]. The quantity of DNA is affected by the size of the available specimen and the proportion of neoplastic tissue remaining for molecular analysis, especially after immunohistochemistry staining is completed for pathologic identifications.
Next-generation sequencing is being evaluated as an option for molecular analysis of many different genes. A recent study looked at 77 colorectal cancer specimens and had KRAS testing performed by both CLIA assay and next-generation sequencing. A 96 % concordant rate between the two assays was found. Additionally 6 % of patients had KRAS mutations that would not usually be tested with standard testing [81]. Given the clinical significance of treatment options and exposure to monoclonal antibodies based on results of KRAS testing, it will be interesting to see if next-generation sequencing will become the gold standard in the future. For now, assays for clinical use will be performed based on the CLIA assay, but it is important to remember the possibility of both false positives and false negatives, which can affect treatment options for patients.
NRAS
NRAS (neuroblastoma RAS viral (v-ras) oncogene homolog) is another member of the Ras family. As with KRAS, NRAS should be tested for in all patients with metastatic colorectal cancer. The same methodologies available for KRAS testing are available for NRAS testing. NRAS codons 12, 13, 59, and 61 should be evaluated, as they are known to predict for poorer response to EGFR inhibitors [80].
The presence of NRAS mutation excludes the use of cetuximab or panitumumab, similar to the presence of KRAS mutations [82]. PCR assays are used to detect NRAS mutations, similar to KRAS.
BRAF
B-Raf proto-oncogene, serine/threonine kinase (BRAF) is an oncogene, like KRAS, that is responsible for MAPK pathway signaling. When BRAF is activated, it leads to constitutive kinase activity and phosphorylation of targets of the RAS/RAF/MAPK signaling pathway [83]. BRAF is mutated in approximately 10 % of colorectal cancers [84]. The most common BRAF mutation is a point mutation substituting a thymidine-to-adenine transversion at nucleotide 1,799 [85]. This results in a single amino acid substitution V600E. KRAS and BRAF mutations are mutually exclusive in colorectal cancer [86] (Figs. 12.13 and 12.14).
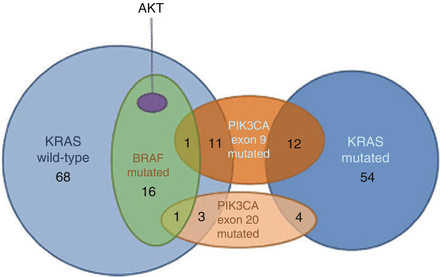
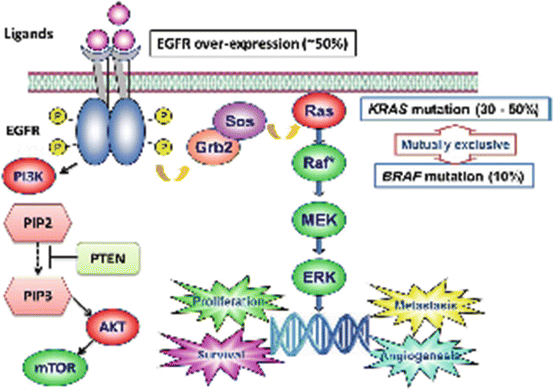
Fig. 12.14
Venn diagram showing the association between AKT exon 3, BRAF exon 15, KRAS exon 2, and PIK3CA exons 9 and 20 mutations expression in 171 samples of primary colorectal cancer [88]
Methodology
The current gold-standard for identifying BRAF V600E mutations are sequencing-based technologies, especially PCR assays. BRAF primers are available [83]. In melanoma, where the use of BRAF inhibitors is approved, the Cobas 4800 BRAF V600 mutation test is approved [89]. Primers for the PCR assay include BRAF codon 600 flanking sequences and allele-specific PCR [90]. QPCR utilizing BRAF V600E mutant-specific probe and BRAF-WT allele-specific probe [91]. A BRAF V600E mutant-specific monoclonal antibody (clone VE1) is now available that can be used on formalin-fixed paraffin-embedded tissue specimens [92]. If immunohistochemistry testing is used to determine BRAF mutation status, only strong to moderately positive staining should be considered positive [93].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


