(1)
Institut Bergonié, Bordeaux, France
Abstract
G-protein-coupled receptors (GPCRs) constitute a huge family of membrane receptors to numerous hormones, neurotransmitters and diverse compounds. A large part of cardiovascular and neurological pharmacology is based upon the knowledge of these receptors and on the identification of molecules able to interfere with them, in an agonistic or antagonistic way. We will only briefly consider this vast domain, so as to focus on some of these receptors, those having a possible link with oncogenesis. In a first part, we will present the overall way of operating of these receptors and the main events resulting from their activation. We will present in a special part a family of GPCR and cognate ligands, which are susceptible to be involved in oncogenesis and metastasis, the chemokine–chemokine receptor families.
As a general feature, the functioning of these receptors involves the activation of heterotrimeric protein complexes called G-proteins, whose α subunit is able to bind and hydrolyse GTP. These proteins activate the membrane enzymes, especially adenylyl cyclase and phospholipase C, which produce second messengers (cyclic AMP, diacylglycerol, inositol trisphosphate, Ca2+). These second messengers ultimately activate various types of serine/threonine kinases, especially protein kinases A (PKAs), B (AKT) and C (PKCs). Heterotrimeric G-proteins may also activate small G-proteins through the activation of their guanine nucleotide exchange factors (GEFs).
6.1 Structure and Mechanism of Action of G-Protein-Coupled Receptors
6.1.1 Receptors
GPCRs are comprised of a single polypeptidic chain equipped with seven transmembrane domains; these domains are made of a series of 22–25 hydrophobic amino acids, which allow the formation of an α-helix inserted within the aliphatic chains on membrane phospholipids. The N-terminal part of the receptors is extracellular while the C-terminal part is intracellular; three loops separating the transmembrane domains are present on each side of the membrane. The polypeptidic chain thus winds in the membrane as a snake, and these receptors are sometimes called ‘serpentine receptors’. The number of receptors in this family is higher than 800, which represents a significant part of the genome; they are divided in several families and subfamilies as a function of their structure, their ligands and the type of binding. With some simplification, one can identify:
The rhodopsin receptors family, the most abundant (about 700 members). Among them are receptors to small molecules with intercellular messenger function: acetylcholine, catecholamines and other biogenic amines; nucleosides and nucleotides; lipid mediators such as retinal, prostaglandins, leukotrienes and thromboxanes, free fatty acids and endocannabinoids; and protein and peptide hormones (corticotropin [ACTH], gonadotropins [FSH, LH], thyrotropin [TSH], oxytocin [OXT], angiotensin [AGT], somatostatin [SST], chemokines, etc.). This family contains also all the olfactory and gustatory receptors, which represent more than 400 different proteins. The binding between receptor and ligand involves deep transmembrane structures of the receptors. Some of these receptors are activated by thrombin- or trypsin-induced proteolysis, which cleaves the N-terminal part of the receptor, which then behaves as an autonomous ligand; they are called protease-activated receptors (PAR).
The secretin receptors family (15 members), which comprises the receptors to various peptide hormones: glucagon, parathormone, calcitonin, and hypothalamic releasing factors (CRH [corticotropin-releasing hormone], GNRH [gonadotropin-releasing hormone] or LHRH [luteinising hormone-releasing hormone], TRH [thyrotropin-releasing hormone]). The binding between ligand and receptor involves the transmembrane domains and the N-terminal part of the receptor.
The glutamate receptors family (22 members), comprising the metabotropic receptors (as opposed to ionotropic receptors, Chap. 15) of several neurotransmitters (glutamate, GABA), which bind to the N-terminal part of the receptor. This family also contains a Ca2+ ‘sensor’ (CASR); the calcium ion binds to an extension of the N-terminal part of the receptor, which then interacts with the deep transmembrane domains and behaves as an ‘auto-ligand’.
The adhesion receptors family, whose receptors have a bulky N-terminal domain allowing them to interact with extracellular molecules, but whose true ligands are mainly proteoglycans.
The frizzled receptors family, which comprises the 10 WNT protein receptors (Chap. 7) and the secondary receptor of the Hedgehog pathway, called smoothened (SMO) (Chap. 9). These receptors present structural analogies with GPCR, but the G-protein activation characteristic for GPCR has not been always demonstrated.
In all cases, ligand–receptor binding induces a conformational change which generates the message. One can consider that the receptors exist under two forms, an active and an inactive form, and that their ligands stabilise one form or the other one. Some receptors have been shown to be activated after dimerisation, but this may not be the case for all receptors.
6.1.2 G-Proteins
The common final way for GPCR activation resides in the involvement of large heterotrimeric G-proteins. All G-proteins are able to bind and hydrolyse GTP (they are therefore GTPases); there is a distinction between ‘small G-proteins’ such as RAS (Chap. 2), RHEB or RAG (Chap. 3) and ‘large heterotrimeric G-proteins’ which are studied here and are transducers of the information received by GPCR. Large heterotrimeric G-proteins are comprised of three polypeptidic chains, called α, β and γ. The β and γ chains are tightly bound in a unique entity βγ. G-proteins are anchored to the membrane by two lipid moieties, the first one being a linear chain (myristoyl or palmitoyl group), which is added post-translationally at the N-terminal extremity of the α subunit, and the second one being a branched chain (farnesyl or geranylgeranyl group), which is post-translationally added to the C-terminal part of the γ subunit. The α subunit contains the binding domain of guanyl nucleotides, which is occupied by GDP at basal state. Large heterotrimeric G-proteins are stably associated with an effector enzyme and can transiently associate with a GPCR.
GPCR ligand binding induces this transient association between a GPCR and a large G-protein, and this binding induces the replacement of GDP by GTP at the level of the α subunit. The α subunit then detaches from the βγ subunit and realises the GTP cleavage thanks to its GTPase activity. During the brief period when the α subunit is bound to GTP, the associated effector enzyme is activated (sometimes inhibited when it has constitutive activity) and can exert (or no longer exert) its catalytic activity. As soon as GTP is hydrolysed, this regulation ceases, the effector enzyme returns to basal state and the α subunit reassociates to the βγ subunit. Similarly, the βγ subunit can activate other effectors before reassociating with the GDP-bound α subunit. Large G-proteins thus function as switches giving one signal only; they must reassociate to a newly activated receptor molecule and exchange GDP against GTP to give a novel impulse to their effectors. Figure 6.1 presents the operating scheme of heterotrimeric G-protein activation.
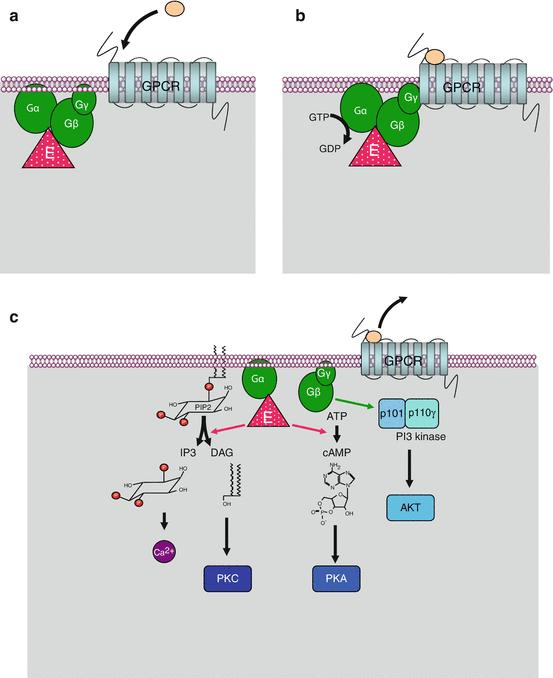
Fig. 6.1
Activation and function of large heterotrimeric G-proteins. At the basal state (a), the 7-transmembrane-domain receptor and the heterotrimeric G-protein complex are dissociated; the α subunit is bound to GDP and to its effector enzyme, adenylyl cyclase (ADCY) or phospholipase C beta (PLCβ). Upon ligand binding (b), receptor and Gαβγ complex are transiently associated; the α subunit exchanges its GDP ligand against GTP. At the active state (c), the GTP-bound α subunit can dissociate from the receptor and the βγ subunit and can activate (or deactivate in the case of an αi subunit) the effector enzyme. cAMP is then generated from ATP by ADCY (or no longer synthesised), and DAG and IP3 are generated by phospholipase C beta (PLCβ), which induces especially protein kinase A (PKA) activation by cAMP, protein kinase C (PKC) activation by DAG and Ca2+ release by IP3. In addition, the βγ subunit is able to activate the regulatory p101 subunit of phosphoinositide 3-kinase gamma (PI3Kγ), which in turn can activate AKT
In front of hundreds of receptors, there is a total of 16 genes encoding the α subunits (genes GNAx), 5 for β subunits (genes GNB1 to GNB5) and 12 for the γ subunits of G-proteins (genes GNG1 to GNG12). The α subunits reflect the tissue and functional diversity of G-proteins, whereas the βγ subunits are rather involved in controlling G-protein activity. There are several types of α subunits, the main ones being αs (gene GNAS), αi (genes GNAI1 to GNAI3), αq (genes GNAQ, GNAL and GNA11) and α12 (gene GNA12 and GNA13), which differ accordingly to the effector enzyme they can activate: GTP-bound αs subunits activate adenylyl cyclases (genes ADCY1 to ADCY10), while αi subunits inhibit this activity; GTP-bound αq subunits activate phospholipases C beta (PLCβ, genes PLCB1 to PLCB4) and GTP-bound α12 subunits activate small G-proteins of the RHO family. Adenylyl cyclase and PLCβ generate second messengers, which are common intermediates of the signals brought by GPCR ligands. The βγ subunit, transiently detached from the α subunit during activation, also has proper effectors, especially the regulatory subunit of PI3 kinase gamma (p101, gene PIK3R5), as well as some types of ionic channels.
The activity of large heterotrimeric G-proteins is regulated by RGS (regulator of G-protein signalling) proteins, which stimulate GTPase activity and are therefore GAPs (GTPase-activating proteins). As a consequence, they deactivate large G-proteins that are brought back to GDP-bound, inactive state. There are about 20 RGS proteins with variable specificity toward G-proteins. Large heterotrimeric G-proteins can also be regulated by phosphorylation: the βγ subunit is able to recruit a kinase (GRK, GPCR kinase) which phosphorylates the activated receptor and accelerates its desensitisation through the formation of a β-arrestin-binding site, inducing receptor internalisation and recycling.
6.2 Second Messengers of GPCR Activation
6.2.1 Adenylyl Cyclases and Cyclic AMP
Adenylyl cyclases (ADCYs) are high-molecular-weight membrane enzymes, comprised of two homologous parts, each made of six transmembrane helices and a large C-terminal domain called the C-domain. The two C-domains are associated and present an ATP-binding site and a catalytic site for its conversion into cAMP. These enzymes are controlled by heterotrimeric large G-proteins; those equipped with an αs subunit activate adenylyl cyclase when bound to GTP, whereas those equipped with an αi subunit inhibit adenylyl cyclase. They are also controlled by multiple factors and integrate much information, in particular through Ca2+ binding and protein kinase C-induced phosphorylation.
Cyclic APM (cAMP) (Fig. 6.2) is the second messenger of a large number of signals received by GPCR: hormones, neurotransmitters, etc. It is involved in the regulation of numerous enzymes, especially in metabolic pathways. cAMP targets are protein kinases A (PKAα, β and γ, genes PRKACA, B and G), which are tetrameric serine/threonine kinases containing two catalytic and two regulatory subunits. They phosphorylate their targets on the serine residues of an RRXS consensus motif. cAMP-induced PKA activation is cooperative, which allows a rapid and wide effect, rapidly terminated by the fact that PKAs phosphorylate and activate cAMP phosphodiesterases (genes PDE4A to PDE4D), which hydrolyse cAMP. PKAs target numerous proteins, among which are glycogen phosphorylase kinase (PHK), which is activated, and glycogen synthase (GYS), which is deactivated, in response to this phosphorylation. PKAs also exert an effect at the gene transcription level, by activating through phosphorylation a transcription factor called CREB (cAMP response element-binding protein), which relocalises into the nucleus and recognises specific promoter sequences of several target genes, called CRE (cAMP response element). cAMP is also able to directly activate EPAC (exchange protein directly activated by cAMP), a GTP–GDP exchange factor of a small G-protein, RAP, which activates other intracellular signalling pathways.
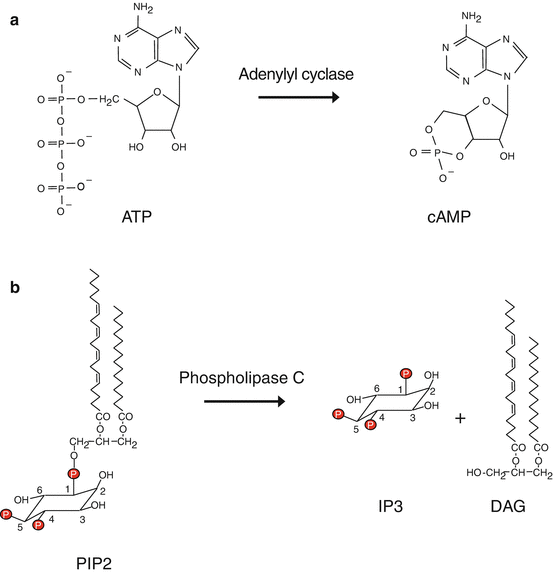
Fig. 6.2
Formation of second messengers. (a) Adenylyl cyclase can convert ATP into cyclic AMP (cAMP). (b) Phospholipase C beta hydrolyses phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol trisphosphate (IP3) and diacylglycerol (DAG)
6.2.2 Phospholipases C, Diacylglycerol and Inositol 1,4,5-Trisphosphate
Phospholipases C (PLCs) are membrane enzymes equipped with a PH (pleckstrin homology) domain allowing them to recognise and bind phosphoinositides. PLCγ can be activated by a tyrosine kinase receptor (TKR, Chap. 1), whereas PLCβs are activated by a GPCR through the action of large heterotrimeric G-proteins equipped with an αq subunit. They act on phosphatidylinositol 4,5-bisphosphate and generate a lipophilic molecule, diacylglycerol (DAG), and a hydrosoluble molecule, inositol 1,4,5-trisphosphate (IP3) (Fig. 6.2). These two molecules are the second messengers of numerous GPCR ligands, hormones and various transmitters.
DAG is the principal physiological activator of protein kinases C (PKCs). This family of nine serine/threonine kinases (genes PRKCx), of variable tissue expression, has numerous substrates with relatively low specificity and exerts various intracellular effects. Before DAG-induced activation, PKCs must be phosphorylated by PDK1 (phosphoinositide-dependent kinase 1), which enables their autophosphorylation on two distinct sites. DAG interaction enables PKC anchoring in the plasma membrane, which is required for catalytic activity. Several other lipid compounds, produced by enzymes other than phospholipase C, such as phospholipases A2 and D and sphingomyelinase, are also able to activate PKCs: lysophosphatidic acid (LPA), arachidonic acid, sphingosine, etc. PKCs are able to bind numerous proteins that direct its action to precise protein targets: STICKs (substrates that interact with C-kinase), such as vinculin (VCN), talin (TLN) and annexin A (ANXA), PICKs (proteins interacting with C-kinase) and RACKs (receptors for activated C-kinase). These proteins play major roles in cell adhesion and polarity and intracellular contacts and explain the multiple actions of PKCs in these processes.
PKCs can be divided in several subfamilies: class A (‘conventional’: PKCα, PKCβ and PKCγ, activated by DAG and Ca2+), class B (‘novel’: PKCδ, PKCε, PKCθ and PKCη) and class C (‘atypical’: PKCι/λ and PKCζ). PKCs phosphorylate many substrates, with variable specificity, depending upon the different members of the family. They are involved in various pathways, especially in cell proliferation, cell cycle entry, cell survival, cell adhesion and cell migration (Fig. 6.3). Among the PKC substrates are the IKK (IκB kinase) proteins, which generate NFκB (Chap. 12), the CRAF (RAF1) MAP3 kinase in the MEK–ERK MAP kinase pathway (Chap. 2), and GSK3β, which is inhibited by this phosphorylation on Ser9 and which is known as a tumour suppressor in the Wnt (Chap. 7) and Hedgehog (Chap. 9) pathways.
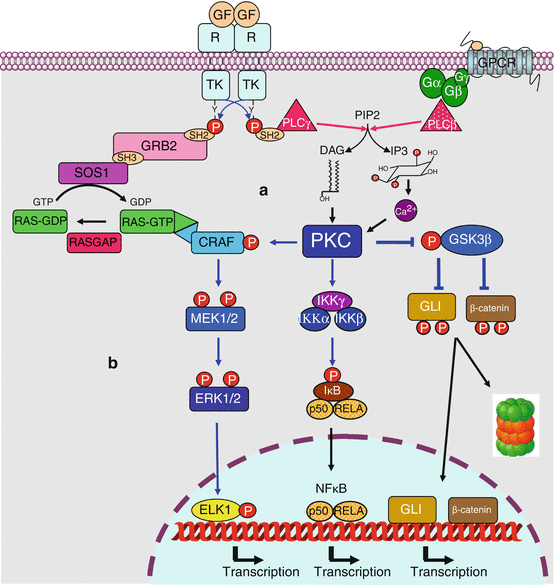
Fig. 6.3




PKC-activating and PKC-activated pathways. (a) PKC-activating pathways. PKCs are activated by diacylglycerol (DAG) and, for some of them, by Ca2+, two second messengers obtained by hydrolysis of phosphatidylinositol 4,5-bisphosphate by a phospholipase C, which can be activated either by tyrosine kinase receptors (PLCγ) or by G-protein-coupled receptors (PLCβ). (b) PKC-activated pathways. Among numerous protein targets, only some of them are indicated. Activating phosphorylation of CRAF and IKK leads to the activation of MAP and NFκB transcription factors, respectively. The phosphorylation of GSK3β is in contrast inhibitory; this kinase phosphorylates GLI (Hedgehog pathway, Chap. 9) and β-catenin (Wnt pathway, Chap. 7), which are transcription factors or transcriptional activators, which leads them to ubiquitinylation and proteasome destruction. This double inhibition explains the activation of these pathways by PKC
Related posts:
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
