Type
Application
Target
Alemtuzumab
Chronic lymphocytic leukemia
CD52
Bevacizumab
Anti-angiogenic therapy
Vascular endothelial growth factor (VEGF)
Cetuximab
Colorectal, head, and neck cancer
Epidermal growth factor receptor (EGFR)
Gemtuzumab
Acute myeloid leukemia
Myeloid cell-surface antigen CD33 on leukemia cells
Ibritumomab
Non-Hodgkin lymphoma
CD20
Nimotuzumab
Squamous cell carcinoma, glioma
EGFR inhibitor
Panitumumab
Colorectal cancer
EFGR
Rituximab
Non-Hodgkin lymphoma
CD20 on B-lymphocytes
Tositumomab
Non-Hodgkin lymphoma
CD20
Trastuzumab
Breast cancer
HER2/neu receptor
Cytokines | ||
|---|---|---|
Interferon-gamma | Melanoma, renal and kidney cancer, follicular lymphoma, hairy cell leukemia | IFN-stimulated gene factor 3 (ISGF3) |
Interlukin-2 | Melanoma, renal and kidney carcinoma, hematological malignancies | Suppressors of cytokine signaling (SOCS) 1, SOCS2, dual-specificity phosphatase (DUSP) 5, DUSP6 |
Short peptides | |
|---|---|
MART-1, gp100, tyrosine, MAGE-3 | Melanoma |
PAP/GM-CSF | Prostate carcinoma |
MAGE-3.A24 | Bladder cancer |
Follicular B-lymphoma | Idiotype/KLH conjugate |
DNA vaccines use recombinant DNA that encodes a specific (defined) antigenic protein. The DNA is incorporated into viruses that are injected directly into patients or, more often, introduced into Dcs obtained from the patients, which are then injected back into them. The DNA expresses the target antigen which triggers or enhances patients’ immune response.
Autochthonous tumor cells (cells taken from the host) have been reintroduced to the host after use of ex vivo techniques (e.g., irradiation, neuraminidase treatment, hapten conjugation, hybridization with other cell lines) to reduce their malignant potential and increase their antigenic activity. Allogeneic tumor cells (cells taken from other patients) have also been used in patients with acute lymphocytic leukemia and acute myeloblastic leukemia.
1.4.2 Nonspecific Immunotherapy
Interferons (IFN-α, -β, -γ) are glycoproteins that have antitumor and antiviral activity. Depending on dose, interferons may either enhance or decrease cellular and humoral immune functions. Interferons also inhibit division and certain synthetic processes in a variety of cells. Clinical trials have indicated that interferons have antitumor activity in various cancers, including hairy cell leukemia, chronic myelocytic leukemia, AIDS-associated Kaposi’s sarcoma, non-Hodgkin lymphoma (NHL), multiple myeloma, and ovarian carcinoma. However, interferons may have significant adverse effects, such as fever, malaise, leukopenia, alopecia, and myalgias.
Certain bacterial adjuvants (BCG and derivatives, killed suspensions of Corynebacterium parvum) have tumoricidal properties. They have been used with or without added tumor antigen to treat a variety of cancers, usually along with intensive chemotherapy or radiation therapy. For example, direct injection of BCG into cancerous tissues has resulted in regression of melanoma and prolongation of disease-free intervals in superficial bladder carcinomas and may help prolong drug-induced remission in acute myeloblastic leukemia, ovarian carcinoma, and NHL.
1.5 Stimulation of Responses In Vivo
The poor immunogenicity of most tumor antigens largely reflects the nonconductive context in which these antigens are naturally presented, as well as tolerance resulting from most tumor antigens being normal proteins aberrantly expressed by the tumor. Therapeutic vaccines have attempted to circumvent these problems by presenting tumor antigens in a more enticing fashion, generally through activated DCs. This has been achieved either by:
Isolating DCs and introducing the antigen ex vivo before returning the DCs to the host
Inoculating dead tumor cells modified to secrete factors such as granulocyte-macrophage colony-stimulating factor (GM-CSF) which promote local accumulation of DCs
Injecting activators of DCs, such as TLR ligands or mAb to CD40 with the antigen
Injecting recombinant vectors that provide both the antigen and a stimulus to the innate immune system [15]
The last category includes plasmid DNA containing the antigen and immunostimulatory CpG sequences as well as recombinant attenuated pathogens, such as adenoviruses or Listeria monocytogenes, that express the antigen and provide TLR ligands to trigger innate responses. However, most vaccinated patients exhibit only weak or undetectable T-cell responses to the tumor antigen and experience no clinical benefit. Therefore, methods to maintain APC activation and sustain immunogenic antigen presentation normally occurring during an encounter with a replicating foreign pathogen will likely be required before vaccines become more predictably beneficial.
An alternative to improving antigen presentation has been to mitigate negative checkpoint signals that limit the T-cell response. Cytotoxic T-lymphocyte antigen-4 (CTLA-4) is a potent negative regulator of T-cell activation. Administration of blocking antibodies to CTLA-4 has had marked effects in murine models and recent clinical trials, with lymphocytic infiltration into tumors and significant antitumor responses, including complete regressions of advanced disease in a fraction of patients [16–18]. However, global in vivo CTLA-4 blockade predictably had effects beyond the antitumor response, causing significant autoimmunity. These studies again demonstrate the potent antitumor activity of T-cells and suggest that learning how to safely and effectively disrupt checkpoint signals should yield substantial therapeutic benefit.
1.6 Adoptive Immunotherapy
There is now an emerging sense that cancer immunotherapy has the potential to effectively cure patients suffering from certain types of cancer. This hope and some of the data that supports one kind of immunotherapy (adoptive cell transfer or ACT) were recently summarized in a review article (Adoptive immunotherapy for cancer: harnessing the T-cell response) [19]. Furthermore, high-dose chemo-radiotherapy followed by rescue from the resulting ablation of normal bone marrow with an allogeneic hematopoietic stem cell transplant (HSCT) has also become standard therapy for many hematologic malignancies. One problem with this treatment is graft-versus-host disease (GVHD), due to allogeneic donor-derived T-cells injuring the “foreign” normal tissues of the host. However, malignant cells that survive chemoradiotherapy are also of host origin, and patients who develop GVHD have lower relapse rates from an associated graft-versus-tumor (GVT) effect. T-cells mediate this antitumor activity, as affirmed by the complete responses sometimes observed in patients who receive infusions of donor T-cells to treat relapse after HSCT and in recipients of a newly developed non-myeloablative allogeneic HSCT regimen in whom, because of the absence of high-dose chemoradiotherapy, all antitumor effects must result from GVT effects [20]. However, the GVT activity with these regimens is often associated with severe and life-threatening GVHD. Ongoing efforts to define antigenic targets with limited tissue distribution, permitting donor lymphocytes to preferentially target malignant cells and not critical normal tissues, coupled with methods to generate and/or select T-cells with such specificities, should provide a much-needed refinement to this approach [21].
An alternative to using allogeneic T-cells to mediate antitumor responses has been to isolate autologous tumor-reactive T-cells, expand the cells in vitro, and then reinfuse the cells back into the patient. This approach circumvents many of the obstacles to generating an adequate response in vivo, as the nature of the APCs and components of the microenvironment can be more precisely controlled in vitro. However, this strategy has required the recent development of methods to extensively manipulate T-cells in vitro with retention of specificity and function, such that after infusion the cells will survive and migrate to and eliminate tumor cells.
Initial therapies used tumor-infiltrating lymphocytes as an enriched source of tumor-reactive cells, but such cells can also usually be obtained from circulating blood lymphocytes. Although optimal methods for stimulating and expanding antigen-specific T-cells in vitro are still being defined, in general, DCs presenting the antigen are used to initially trigger reactive T-cells, which can then be selected and stimulated with antibodies to CD3. Supplemental cytokines are provided during cell culture to support lymphocyte proliferation, survival, and differentiation. With this approach, it has been possible to expand tumor-reactive T-cells to enormous numbers in vitro, infuse billions of specific cells without overt toxicity to achieve in vivo frequencies beyond that attainable with current vaccine regimens, and mediate regression and occasionally complete elimination of large disseminated tumor masses. However, despite the high in vivo frequencies of tumor-reactive effector cells achieved, only a fraction of patients respond, indicating the existence of additional hurdles. One essential requirement is that infused cells must persist to mediate an effective response. Analogous adoptive therapy trials for cytomegalovirus and Epstein-Barr virus infection in immunosuppressed hosts have demonstrated increased in vivo proliferation and persistence of CD8+ effector T-cells in the presence of specific CD4+ helper T-cells [22]. Such CD4+ T-cells likely provide many beneficial functions, including cytokine production and APC activation, which can improve the quality and quantity of the CD8+ cell responses, as well as direct effector activities against infected or tumor targets. However, unlike viral responses that induce robust CD4+ and CD8+ responses, identifying and characterizing the specificity of tumor-reactive CD4+ T-cells has proven considerably more difficult than with CD8 responses. Additionally, obstacles to safely maintaining a CD4+ response reactive with a potentially normal protein remain to be elucidated. Consequently, CD4 help is largely provided to transfer tumor-reactive CD8 cells in the form of surrogate exogenous cytokines. The largest experience is with IL-2, which prolongs persistence and enhances the antitumor activity of transferred CD8+ cells [23]. Alternative cytokines such as IL-15, IL-7, and IL-21, as well as activation of APCs with antibodies to CD40, are currently being evaluated in preclinical studies.
The infusion of T-cell clones, rather than polyclonal T-cell lines, represents an appealing refinement of adoptive therapy, because the specificity, avidity, and effector functions of infused cells can be precisely defined (Fig. 1.1). This facilitates subsequent analysis of requirements for efficacy, basis for toxicity, and rational design of improved therapies. The transfer of antigen-specific CD8+ T-cell clones has been shown to be effective for prevention of viral infections and treatment of malignant disease. Such studies have also formally demonstrated that low, nontoxic doses of IL-2 are sufficient to promote the in vivo persistence and antitumor activity of CD8+ T-cells.
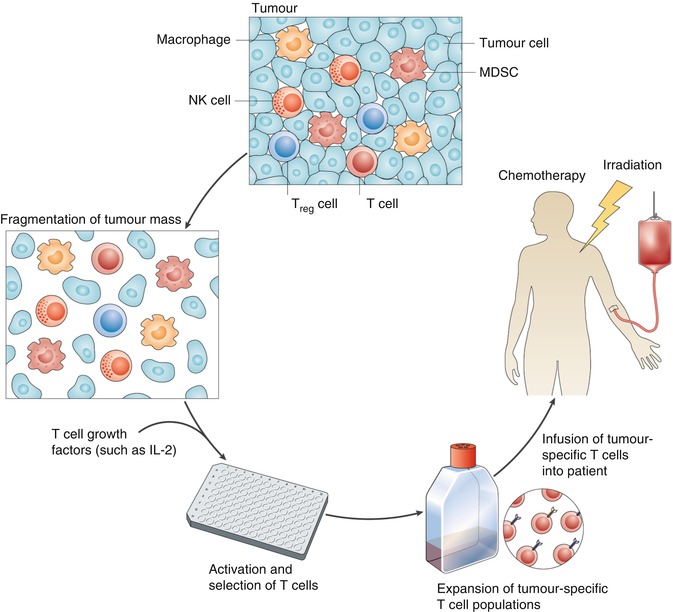
Fig. 1.1
Tumors are often complex masses containing diverse cell types. These masses can be surgically resected and fragmented, and the cells can be placed in wells into which a T-cell growth factor, such as interleukin-2 (IL-2), is added. T-cell populations that have the desired T-cell receptor (TCR) specificity can be selected and expanded and then adoptively transferred into patients with cancer. Prior to this adoptive transfer, hosts can be immunodepleted by either chemotherapy alone or chemotherapy in combination with total-body irradiation. The combination of a lymphodepleting preparative regimen, adoptive cell transfer, and a T-cell growth factor (such as IL-2) can lead to prolonged tumor eradication in patients with metastatic melanoma. MDSC myeloid-derived suppressor cell, NK natural killer, Treg regulatory T (Reprinted by permission from Nature Publishing Group: Restifo et al. [19])
1.7 Cancer Vaccines
Therapeutic cancer vaccines target the cellular arm of the immune system to initiate a cytotoxic T-lymphocyte response against tumor-associated antigens [24]. The development of human therapeutic cancer vaccines has come a long way since the discovery of MHC-restricted tumor antigens in the 1980s. The simplest model of immune cell-mediated antigen-specific tumor rejection consists of three elements: appropriate antigen, specific for the tumor, efficient antigen presentation, and the generation of potent effector cells. Moreover, the critical time when immune responses against the tumor are most important should also be determined. While eliminating some early transformed cells may be ongoing in an asymptomatic way as part of the immunosurveillance, if early elimination failed, equilibrium between small tumors and the immune system may be established. If the immune system is unable to maintain this equilibrium, tumors may escape and it is this last phase when they become symptomatic. Therapeutic cancer vaccines are applied in this last phase in order to reverse the lack of tumor control by the immune system. In addition to the increasing knowledge about how to optimize the elements of antitumor immunity in order to generate clinically relevant responses, there is an ever-increasing list of immune evasion mechanisms impeding the efforts of cancer vaccines. This indicates that the elements necessary for immune-mediated tumor rejection need to be optimized [25].
Potential tumor-associated antigens (TAAs) can be identified by the elution of peptides from MHC molecules on tumor cells [26] or with proteomic approaches such as two-dimensional gel electrophoresis, MALDI-MS and SELDI-MS (matrix-assisted or surface enhanced laser-desorption ionization mass spectrometry) [27]. Serological analysis of recombinant cDNA expression libraries (SEREX) is another widely used method; it utilizes sera of cancer patients to detect over expressed antigens from tumor cDNA libraries [28]. Furthermore, several RNA-based methods have also gained importance: transcriptome analysis that include DNA microarrays [29], serial analysis of gene expression (SAGE) [30], comparative genomic hybridization (CGH) [31], and massively parallel signature sequencing (MPSS) [32]. These methods provide an enormous amount of information and require complex computer-aided analysis and interpretation of the data, referred to as gene expression profiling. This is necessary in order to find gene expression patterns and to distinguish them from noise [33].
Following promising in vitro immunogenicity studies [34], multicenter vaccine trials have been organized with the sponsorship of the Cancer Vaccine Collaborative (NCI and Ludwig Institute for Cancer Research). These trials have provided some information about the optimum route of administration, type of vaccine, type of adjuvant, endpoints, etc. [35]. When testing the immunogenicity of candidate antigens and defining epitopes, it should be remembered that T-cells with high avidity for self-antigen undergo negative selection during T-cell development; thus, the new TAAs may only generate T-cell responses of intermediate or low affinity. Furthermore, the wide range of restriction elements in the human population means that due to the combination of tolerance and immunodominance, potentially ideal TAAs will not be equally immunogenic in all patients. Antigen loss may also occur during tumor progression, as TAAs which are not necessary for the maintenance of the transformed phenotype may be deleted and tumor cells in advanced disease may express antigens different from those in early stages [36]. Another promising approach to break this immune tolerance consists of the application of anti-idiotypic (anti-Id) mAbs, so called Ab2, as antigen surrogates. This vaccination strategy also allows immunization against nonprotein antigens (such as carbohydrates). In some clinical studies, anti-Id cancer vaccines induced efficient humoral and/or cellular immune responses associated with clinical benefit (see review by Ladjemi 2012) [37].
1.7.1 Dendritic Cells
DCs are the main antigen-presenting cells in the body [38], and their generation for antitumor immunity has been the focus of a vast array of scientific and clinical studies [39]. They are the main antigen-presenting cells (APCs) in the body. Immature DC (iDC) patrols the peripheral tissues, sampling antigen from the environment. Following their activation, DCs undergo a maturation process that involves the upregulation of T-cell co-stimulatory molecules (e.g., CD80, CD86), increased cytokine secretion, a transient increase in phagocytosis followed by reduced antigen uptake, and expression of migratory molecules such as CCR7. These changes equip mature DC (mDC) to prime naive T-cells in the lymph nodes, in contrast to iDC that induce T-cell tolerance to antigen [40].
The ability of DCs to present protein tumor antigens (T-Ags) to CD4+ and CD8+ T-cells is pivotal to the success of therapeutic cancer vaccines. DCs specialized capacity to cross-present exogenous Ags onto MHC class I molecules for generating T-Ag-specific cytotoxic T lymphocytes (CTLs) has made these cells the focal point of vaccine-based immunotherapy of cancer (Fig. 1.2).
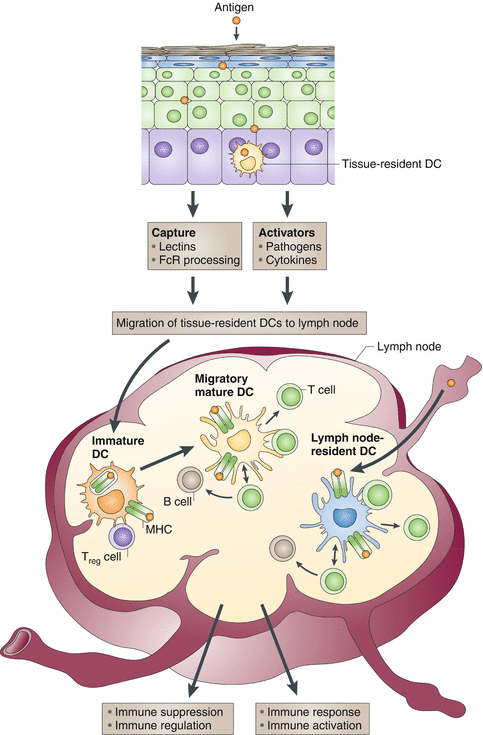
Fig. 1.2
Antigens can reach lymph nodes through two pathways: via lymphatics, where the antigen is captured by lymph node-resident dendritic cells (DCs), or via tissue-resident DCs. These immature DCs capture antigens, and DC activation triggers their migration toward secondary lymphoid organs and their maturation. DCs display antigens in the context of classical major histocompatibility (MHC) class I and MHC class II molecules or in the context of nonclassical CD1 molecules, which allow the selection of rare antigen-specific T-lymphocytes. Activated T cells drive DCs toward their terminal maturation, which induces further expansion and differentiation of T lymphocytes into effector T cells. If DCs do not receive maturation signals, they will remain immature and antigen presentation will lead to immune regulation and/or suppression. Treg cell, regulatory T-cell (Reprinted by permission from Nature Publishing Group: Palucka and Banchereau [136])
Dendritic cells can be loaded exogenously with TAA using whole cell populations or short peptides corresponding to epitopes from specific TAA. While the use of DC pulsed with short peptides can yield information on immune activation following therapy, they are not ideal therapeutic agents for a number of reasons. The most obvious reason is that the use of specific TAA depends on the identification of relevant TAA and not all cancers have well-defined TAA. Moreover, TAA expression within a tumor can be very heterogeneous [41]; thus, priming CTL specific for defined TAA peptides may encourage the outgrowth of non-expressing clones, leading to immune evasion. Furthermore, both MHC-1 and MHC-II epitopes are required for efficient T-cell priming. While a number of MHC-1-restricted peptides have been identified, fewer MHC-II epitopes are known. Synthetic long peptides, comprising both MHC-I and MHC-II epitopes, which require processing by DC before presentation, can overcome some of the limitations of small peptides, as they lead to extended epitope presentation.
An alternative to pulsing with peptide epitopes is to load DC with whole tumor cell preparations in the form of lysates or whole dead cells or by fusing DC with tumor cells [42]. Both allogeneic and autologous tumor material have been used to load DC with clinical trials carried out using preparations using both types [43].
Genetic modification of DC, using recombinant DNA viruses encoding TAA, has been demonstrated by several groups and can enhance T-cell priming potential via antigen presentation. DCs transduced to express the model tumor antigen β-galactosidase, using a recombinant adenoviral vector, were able to generate antigen-specific CTL responses [44]. A phase I/II trial using genetically modified DC showed that autologous DC could be transduced with high efficiency using a replication-defective adenovirus expressing full length melanoma-associated antigen recognized by T-cells (MART-1) and that the DC processed and presented the antigen for at least 10 days. Evidence of MART-1-specific CD4+ and CD8+ responses was found in around 50 % of patients following vaccination [45].
In addition to loading DC with antigen, genetic approaches have been used to further optimize the maturation state of DC, for example, DC transfected with GM-CSF demonstrated increased antigen presentation and better migratory capacity, which translated into enhanced immune priming in vivo [46]. Other approaches include genetically modifying DC using adenoviral or retroviral vectors to directly express TH1 cytokine IL-12 [47], an adenovirus encoding CD40L [48] and modifying DC to express co-stimulatory molecules CD40L, CD70, and TLR4 called “TriMix” [49] and heat shock protein [50]. Furthermore, vaccines coupled to TLR ligands lead to efficient CTl activation by endogenous DC [51], and the use of oncolytic viruses also looks particularly promising [52].
Despite the use of mature DCs in vaccination trials, results from multiple clinical trials with DC-based vaccines have been contradictory, and only fractions of enrolled patients show potent antitumor or antiviral immune responses with moderate clinical response rates (approximately 10–15 %) (see reviews [53, 54]). Several studies suggested that this is because of inefficient activation of Th1-polarized responses due to incomplete DC maturation. As a result, different strategies are currently being pursued in order to improve the efficacy and outcome of DC-based cancer vaccines. Considering the aforementioned powerful immune-stimulatory properties possessed by IL-12p70, DC-based vaccination strategies may consistently benefit from incorporation or endogenous induction of this cytokine. In a first phase I clinical trial by the group of Czerniecki [55], 13 breast cancer subjects were injected intranodally with short-term DCs activated with a cytokine-cocktail consisting of IFN-γ and LPS in order to induce IL-12p70-secreting DCs. The authors reported induction of robust detectable immunity as evidenced by in vitro monitoring of circulating vaccine-induced antigen-specific CD4+ and CD8+ T-cells, as well as both T-and B-cell infiltrates into tumor region and dramatic reductions in tumor volume. Moreover, it has been demonstrated by others that DCs electroporated with mRNA encoding CD40 ligand, CD70, and constitutively active toll-like receptor 4, so-called TriMix DCs, display increased potential for the induction and amplification of tumor-specific responses in patients with advanced melanoma [56, 57].
One of the major obstacles against successful DC vaccination is the immunosuppressive mechanisms triggered by the tumor cells. Under the influence of the tumorigenic microenvironment, the host DCs may acquire a tolerogenic phenotype. These tumor-conditioned DCs could, in return, produce a variety of immunosuppressive molecules, thus further supporting tumor immune escape [58]. With respect to tackling different arms of the immune system, many different approaches are currently being pursued. In particular, considering the distinct ability of different DC subsets in inducing both innate and adaptive immunity, the exploitation of specific subsets of DCs to elicit the desired immune response is anticipated. Although pDCs primarily contribute to innate antiviral immune responses by producing IFN-α/β, this ability has also been reported to activate other DCs, including those involved in cross-priming and consequently greater activation of adaptive immune responses. In so doing, pDCs may play a critical role in provoking cancer immunity. Therefore, combination therapies aiming at interaction of pDCs and cDCs to stimulate T-cell priming and hence effective antitumor or antiviral immunity are needed in cancer patients and chronically infected patients.
1.7.2 Physical Barriers, Tumor Stroma and Vessels
The tumor environment represents another challenge for cancer vaccines. Established epithelial tumors can be surrounded by basal-membrane-like structures which prevent infiltration by lymphocytes and the expansion of tumor-specific T-cells at the tumor site and in lymphoid tissues [59]. Solid tumors larger than about 1–2 mm in diameter require the presence and support of stromal cells for blood supply, growth factors, and structural support. The stroma consists of cancer-associated fibroblasts (CAF), tumor endothelial cells (TEC), and tumor-associated macrophages (TAM) and can represent more than 50 % of the tumor tissue depending on the type tumor [60]. Stromal cells do not only represent a physical barrier but also release soluble mediators (TGF-β, IL-10, prostaglandin) which inhibit immune responses and promote angiogenesis and tumor progression [61, 62]. Conventional cancer treatments, such as de-bulking surgery, chemotherapy, or radiotherapy, not only destroy tumor cells but also destroy or damage stromal cells that may contribute to breaking immunological resistance and immunosuppression [63]. The intricate interplay between tumor and stroma attracts their simultaneous immune destruction: when highly expressed TAAs on tumor cells are cross-presented by stromal cells to T-cells, the stromal component also becomes a target of cytotoxic T-cell killing [64].
TGFβ-1 regulates the production of cytokines and growth factors by stromal and tumor cells, such as fibroblast growth factor (FGF), connective tissue growth factor (CTGF), and vascular endothelial growth factor (VEGF), which promote angiogenesis and tumor progression. The new tumor vasculature is generally both structurally and functionally abnormal, which makes trafficking/recirculation of the tumor tissue by lymphocytes and treatments including cancer vaccines, extremely difficult. Anti-angiogenic treatments, including immunological targeting of antigens overexpressed on endothelial cells during angiogenesis or antibody blockade of VEGF-receptors, “normalize” the tumor vasculature [65, 66]. This treatment also reverts epithelial tumors to noninvasive type and may also aid the penetration of vaccines and other treatments in the tumor tissue. Moreover, IL-12 inhibits angiogenesis via an IFN-γ-mediated pathway [67], while adoptively transferred tumor-specific CD8+ T-cells destroy the vasculature of established tumors via an antigen-independent, IFN-γ-dependent mechanism [68].
1.8 Mechanisms of Tumor-Induced Tolerance/Escape from the Immune System
Despite the evidence that immune effectors play a significant role in controlling role in tumor growth under natural conditions or in response to therapeutic manipulation, it is well known that malignant cells can evade immunosurveillance [69]. This is in part due to the fact that peptides with sufficient immunogenic potential are not presented by malignant cells to antigen-presenting cells under molecular/cellular conditions conducive to an effective immune response. From a Darwinian perspective, the neoplastic tissue can be envisaged as a microenvironment that selects for better growth and resistance to the immune attack. Cancer cells are genetically unstable and can lose their antigens by mutation. This instability, combined with an immunological pressure, could allow for selective growth of antigen-loss mutants [70]. Mechanistically this could operate at several levels including loss of the whole protein or changes in immunodominant T-cell epitopes that alter T-cell recognition, antigen processing, or binding to the MHC. Antigen loss has been demonstrated in patients with melanoma and B-cell lymphoproliferative disease [71, 72]. Moreover, many cancer vaccines aim to induce a therapeutic CD8+ cytotoxic T-cell response against TAAs. This in turn is dependent on correct processing and presentation of TAAs by MHC class I molecules on tumor cells. This pathway is complex and involves multiple intracellular components. Defects in the components of the MHC class I antigen processing pathway are frequently found in human cancers and can occur in concert with the loss of tumor antigens [73, 74].
Other cancer-related mechanisms underlying tumor immune escape include loss of TAA expression [3], lack of co-stimulatory molecules expression [75], inactivating mutations of antigen presentation-related molecules [76], and production of soluble immunosuppressive factors, e.g., transforming growth factor beta (TGF-β), IL-10, reactive oxygen species (ROS), and nitric oxide (NO), produced by tumor cells. Furthermore, tumor-infiltrating immune cells such as suppressor immune cells, e.g., T regulatory (Treg) cells, macrophages, and myeloid-derived suppressor cells (MDSC), also influence this phenomenon and are now discussed in more detail.
1.8.1 Treg Cells
Since their discovery in the 1960s as suppressive T-cells, Tregs have been extensively studied in a wide range of both physiological and pathological conditions in human [77]. Treg suppress T-cell responses and provide another mechanism compromising the development of effective tumor immunity [78]. These cells are usually CD4+ and are distinguishable phenotypically by expression of CD25 (the chain of the IL-2 receptor required for high affinity binding), high levels of CTLA-4, the glucocorticoid-induced TNF-related receptor (GITR), and the forkhead transcription factor Foxp3. Treg cells can arise in response to persistent antigen stimulation in the absence of inflammatory signals, particularly in the presence of TGF-ß, and have been detected in increased frequency in some cancer patients. Furthermore, tumor-induced expansion of regulatory T-cells by conversion of CD4+ CD25+ lymphocytes is thymus and proliferation-independent [79]. Thus, depleting Treg cells in vivo may facilitate the elaboration of effective antitumor T-cell responses.
Inhibiting Treg cell function in patients with cancer is an essential step if new therapies, especially immunotherapies, are to be clinically successful. Initial studies have indicated that depleting Treg cells from cancer patients might be a valid approach; more recent preliminary data has raised the hypothesis that functionally inactivating Treg cells might be a better alternative. Studies in murine tumor models targeting all CD25+ T-cells for depletion have appeared promising [80]. However, activated effector CD8+ and CD4+ T-cells also express CD25, and depletion of these cells during the acute phase of the antitumor T-cell response may severely limit the application of this approach. The availability of the anti-CD25 mAb, PC61, has enabled the effects of Treg-cell depletion to be tested in murine models [81]. Despite some efficacy, intrinsic limitations apply when PC61 is used to treat established tumors as time course experiments have reported that its efficacy is lost as tumors progress [82]. Other mAbs to human CD25 that are available for clinical use, such as daclizumab, block IL-2 and receptor interactions are used to treat hematologic malignancies [83]. However, to date, most studies in humans have used the immunotoxin denileukin difitox (Ontak), a fusion protein between the IL-2 and diphtheria toxin, to selectively kill lymphocytes expressing the IL-2 receptor. The in vivo antitumor efficacy is still under preclinical and clinical investigation, and discrepant results have been reported so far.
Another approach is to inhibit tumor-specific Treg-cell expansion which could be achieved by inhibiting the indoleamine 2, 3-dioxygenase (IDO) pathway. Preclinical data confirm that the administration of an IDO inhibitor significantly decreases the rate of peripheral conversion and dramatically impairs tumor growth [84]. Another possible target is transformed growth factor (TGF), involved in both proliferation and conversion of Treg cells in tumor bearers. Genetically engineered mice that express a dominant negative form of the TGF receptor on lymphocytes show reduced, if not absent, growth of several transplanted tumors [85]. Moreover, CTLA-4 blockade or GITR triggering has been shown to reverse immune suppression as a result of Treg function both in vitro and in vivo [86].
Ultimately, by inducing Treg expansion, the tumor takes advantage of the inhibitory function that these cells exert on all the immune components. Avoiding the physical elimination of Treg cells would be potentially useful as it would prevent the induction of a new wave of peripherally converted Treg cells that are endowed with a wide TCR repertoire. Conversion would also redirect potential effector T-cells toward the Treg-cell phenotype. Alternatively, Treg-cell inactivation is a suitable strategy, which would functionally impair Treg-cell suppression without changing the TCR repertoire of the expanded Treg-cell population. Triggering of TLR8 or OX40, and potentially blocking adenosine, might improve the chances of neutralizing Treg-cell immunosuppression in cancer immunotherapy.
1.8.2 Myeloid-Derived Suppressor Cells
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of cells that expand during cancer, inflammation, and infection and have a remarkable ability to suppress T-cell responses [87]. Although suppressive myeloid cells were described more than 20 years ago in patients with cancer [88], their functional importance in the immune system has only recently been appreciated.
Accumulating evidence has now shown that that this population of cells contributes to the negative regulation of immune responses during cancer and other diseases. Common features to all MDSCs are their myeloid origin, their immature state, and a remarkable ability to suppress T-cell responses. In addition to their suppressive effects on adaptive immune responses, MDSCs have also been reported to regulate innate immune responses by modulating the cytokine production of macrophages [89]. Studies have shown that the expansion and activation of MDSCs are influenced by several different factors, which can be divided into two main groups. The first includes factors that are produced primarily by tumor cells, which promote the expansion of MDSCs through the stimulation of myelopoiesis and inhibit the differentiation of mature myeloid cells. The second group of factors is produced mainly by activated T-cells and tumor cells and is involved in directly activating MDSCs. It has also become clear that the suppressive activity of MDSCs requires not only factors that promote their expansion but also factors that induce activation. The expression of these factors, which are produced mainly by activated T-cells and tumor stromal cells, is induced by different bacterial and viral products or as a result of tumor cell death [90].
The immunosuppressive activities of MSDCs require direct cell-cell contact, suggesting that they function either through cell-surface receptors and/or through short-lived soluble mediator. Such mediators include arginase and nitric oxide synthase (iNOS) [91], reactive oxygen species (ROS) [92], peroxynitrite [93]. Moreover, it has been reported that MDSCs promote de novo development of the FOXP3+ Treg cells in vivo [94]. As they are one of the main immunosuppressive factors in cancer and other pathological conditions, several different therapeutic strategies that target these cells are currently being explored. These include promoting myeloid-cell proliferation [95], inhibition of MDSC expansion [96], inhibition of MDSC function [97], and elimination of MDSC [98]. Ultimately, the roles of specific MDSC subsets in mediating T-cell suppression, and the molecular mechanisms responsible for the inhibition of myeloid differentiation, need to be elucidated. The issue of whether T-cell suppression occurs in an antigen-specific manner remains to be clarified, as do the mechanisms that induce MDSC migration to peripheral lymphoid organs. Some of the main priorities in this field should include a better characterization of human MDSCs and a clear understanding of whether targeting these cells in patients with various pathological conditions will be of clinical importance.
Related posts:
 Novel Strategy of Cancer Immunotherapy: Spiraling Up
Novel Strategy of Cancer Immunotherapy: Spiraling Up
 Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
 Polarization of Tumor Milieu: Therapeutic Implications
Polarization of Tumor Milieu: Therapeutic Implications
 Photodynamic Therapy and Antitumor Immune Response
Photodynamic Therapy and Antitumor Immune Response
 Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


