Agent
Molecular targets
Phase (comparator)
Patient population
Clinical activity in RCC
References
Tivozanib (AV-951)
VEGFR-1, 2, 3
Phase III (TIVO-1) (vs. sorafenib)
Clear cell RCC
PFS (11.9 vs. 9.1 mo)
[13]
More than one prior systemic therapy (does not include targeted therapy)
OS (28.8 vs. 29.3 mo)
ORR (33 vs. 23 %)
Cediranib (AZD2171)
VEGFRs, c-kit, PDGFRβ, FGFR1
Phase II (vs. placebo)
Metastatic or recurrent clear cell RCC
PFS (12.1 vs. 2.8 mo)
[16]
ORR (34 vs. 0 %)
Dovitinib (TKI258)
FGFR1, 2, 3, VEGFR-1, 2, 3
Phase II (single arm)
Advanced RCC treated with at least a VEGFR inhibitor and a mTOR inhibitor
PFS (3.7 mo)
[20]
OS (11.8 mo)
Regorafenib (BAY73–4506)
VEGFR-1, 2, 3, PDGFRβ, FGFR1, Tie2
Phase II (single arm)
Previously untreated advanced RCC (Clear cell RCC)
PFS (11.0 mo)
[25]
ORR (39.6 %)
Cabozantinib
MET, VEGFR-2
Phase II (single arm)
Advanced RCC, at least one or two lines of prior systemic therapy
PFS (14.7 mo)
[29]
RET, c-kit, AXL, FLT3
OS (not yet reached)
ORR (28 %)
Tivantinib (ARQ 197)
MET
Phase I (single arm)
Advanced RCC
Clinical benefit (60 %)
[33]
Foretinib (XL880)
MET, VEGFRs, RON, AXL, Tie2
Phase II (single arm)
Advanced RCC
PFS (9.3 mo)
[34]
No more than one prior systemic therapy
OS (not yet reached)
ORR (13.5 %)
Other strategies for developing new RTK inhibitors are inhibiting other pathways, except for the VEGF pathway, which relates to the RCC biology, or inhibiting pathways that are activated when RCCs develop resistance to VEGFR-targeted therapy.
In addition, inhibition of the VEGF pathway is reportedly less effective in non-clear cell RCC, including papillary RCC, than in clear cell RCC [9]. Therefore, different strategies are needed to treat non-clear cell RCC. For instance, MET inhibitors such as tivantinib and foretinib were developed because papillary RCC is frequently associated with upregulation of the c-MET pathway [10].
14.2.1 Tivozanib (AV-951)
TKIs which have a high affinity for the target molecule may decrease their off-target effects. Tivozanib is a quinoline-urea derivative TKI, a potent VEGFR TKI with high activity against VEGFR-2, and strongly inhibits VEGFR-1 and VEGFR-3 [11, 12]. Tivozanib blocks the VEGFR-1, VEGFR-2, and VEGFR-3 tyrosine kinases at picomolar concentrations [11]. In the Phase II randomized discontinuation trial, 272 patients with mRCC received open-label tivozanib 1.5 mg/day (3 weeks on followed by 1 week off) for 16 weeks [13]. Most of the primary renal tumors revealed a clear cell histology (83 %). In the overall study population, the ORR was 24 % and the median PFS was 11.7 months with tivozanib treatment. After 16 weeks of tivozanib treatment, the 118 patients with a stable disease (SD) were randomly assigned to a placebo or tivozanib group. The median PFS was 10.3 months in the tivozanib group and 3.3 months in the placebo group. The common AEs were hypertension, dysphonia, diarrhea, and asthenia. Among these, hypertension was the most common (all grade; 45 %). TIVO-1 trial was an open-label, randomized Phase III trial for patients with clear cell RCC and no more than one prior systemic therapy that could not include VEGF- or mTOR-targeted therapies [14]. A total of 517 patients were randomly assigned to either a tivozanib (1.5 mg/day for 3-week-on 1-week-off schedule) or sorafenib (400 mg twice daily) group. In the overall population, PFS was significantly longer in patients receiving tivozanib (11.9 months) than in those receiving sorafenib (9.1 months; hazard ratio = 0.797; 95 % CI, 0.639–0.993; p = 0.042). The overall response rate (ORR) favored tivozanib, which was 33 % for tivozanib and 23 % for sorafenib. The final overall survival (OS) was not significantly different between sorafenib and tivozanib (median, 29.3 vs. 28.8 months, p = 0.105), and the tendency for a longer OS was observed in the sorafenib group. A greater proportion of patients in the sorafenib group received the next-line targeted RCC treatment (63 %), whereas 13 % of the patients in the tivozanib group received the next-line treatment. Of the patients who received sorafenib, 61 % (156 patients) switched to tivozanib. With regard to AEs, hypertension (44 % vs. 34 %) was more common with tivozanib and dysphonia with sorafenib (21 % vs. 5 %). Reactions involving the skin of the hand or foot (54 % vs. 14 %) and diarrhea (33 % vs. 23 %) were more common with sorafenib than with tivozanib.
14.2.2 Cediranib (AZD2171)
Cediranib is an oral VEGFR TKI that has an affinity for VEGFRs, c-kit, PDGFRβ, FGFR-1, and several other kinases [15]. Cediranib inhibits VEGFR-2 tyrosine kinase activity at a subnanomolar concentration and VEGFR-1 and VEGFR-3 tyrosine kinase activity at a nanomolar concentration [16]. A Phase II, randomized, double-blind trial was performed that compared the efficacy of cediranib with a placebo in patients with metastatic or recurrent clear cell RCC [17]. The patients had not previously received a VEGFR inhibitor. Patients in the placebo group could cross over to open-label cediranib at 12 weeks or earlier if their disease had progressed. The cediranib group included 53 patients, whereas the placebo group included 18 patients. After 12 weeks of therapy, a significant difference was observed in the mean percent change in tumor size from baseline (primary end point) between the cediranib and placebo groups (20 % reduction vs. 20 % increase). PR was achieved in 34 % of patients treated with cediranib and 47 % of the patients experienced SD. Of the 18 responders, 11 (61 %) had responses lasting for more than 1 year. Cediranib treatment significantly prolonged PFS compared with the placebo (median PFS 12.1 vs. 2.8 months, p = 0.017). The most common AEs in patients treated with cediranib were diarrhea, hypertension, fatigue, and dysphonia. Cediranib monotherapy demonstrated clinical activity in patients with advanced RCC with an acceptable AE profile.
14.2.3 Dovitinib (TKI258)
Tumor escape from anti-VEGF therapy may include various signaling pathways, including the fibroblast growth factor (FGF) signaling pathway. FGF signaling is one of several pathways responsible for tumor resistance after inhibition of the VEGF pathway [18]. Dovitinib is a potent inhibitor of FGFRs (1, 2, and 3) and 3 VEGF receptors [19]. In mouse xenograft models (Caki-1 cells), dovitinib treatment showed an 83 % reduction in tumor volume, whereas sunitinib and sorafenib treatments showed a 66 % and 16 % reduction, respectively [20]. The Phase I trial for dovitinib was performed for mRCC pretreated with VEGFR inhibitor, mTOR inhibitors, cytokine therapy, or a combination of these treatments [20]. A maximum tolerated dose was determined as 500 mg dovitinib (5-day-on 2-day-off schedule). The most common AEs were nausea, vomiting, diarrhea, and asthenia. In the Phase II trials, 67 patients with advanced RCC were treated with dovitinib (500 mg/day) [21]. The patients were previously treated with at least a VEGFR inhibitor and an mTOR inhibitor. Patients with RCC who were previously treated with mTOR and VEGFR inhibitors showed high baseline FGF levels. The elevated baseline FGF levels after VEGF inhibition and the cross talk between FGF and VEGFR signaling led us to consider that a strategy that simultaneously targeted both signaling pathways is possibly useful. The ORR and disease control rate 8 weeks after the treatment were 1.8 % and 52.7 %, respectively. The median PFS and OS were 3.7 and 11.8 months, respectively. Dovitinib treatment resulted in an increased circulating FGF23 level, indicating inhibition of FGFR1. In addition, there were significant decreases in soluble VEGFR-2, and modest increases in VEGF and placental growth factor. The most frequent AEs for all grades were nausea, diarrhea, vomiting, decreased appetite, and fatigue. The GOLD trial ( ClinicalTrials.gov ., NCT01223027) is an ongoing Phase III trial that compares dovitinib with sorafenib in patients with advanced RCC after failure of one VEGF inhibitor and one mTOR inhibitor.
14.2.4 Regorafenib (BAY73–4506)
Regorafenib is a potent inhibitor of several angiogenic RTKs, including VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-β, FGFR-1, and Tie2. In addition, regorafenib inhibits various oncogenic RTKs (c-KIT and RET) and intracellular signaling kinases such as RAF [22]. A unique characteristic of regorafenib is that it inhibits Tie2. The angiopoietins are an important pro-angiogenic growth factor family that signals through the Tie2 receptor [23, 24]. Tie2 is not found in normal vasculature but found only in tumor vascular endothelial cells [25].
A Phase II open-label, nonrandomized study was conducted with 49 patients with advanced RCC [26]. Patients with previously untreated metastatic or unresectable clear cell RCC received regorafenib (160 mg per day, 3 week-on 1-week-off schedule). The tumor response was assessable for 48 patients and 19 patients (39.6 %) showed an objective response (PRs in all patients). Ten of the 19 partial responders maintained a response for more than 12 months and 5 had a response for more than 18 months. Tumor shrinkage occurred in 39 patients (81 %) with 25 patients (52 %) showing a shrinkage of at least 30 %. A clinical benefit (CR, PR, or SD) was noted in 39 patients (81 %; median PFS was 11.0 months). Patients who showed greater increases in plasma concentration of CK18M30 were more likely to show a tumor shrinkage of at least 40 %. Baseline concentrations of soluble Tie1 and TIMP2 tended to be higher in the group that achieved at least 40 % maximum tumor shrinkage. AEs occurred in 48 patients (98 %) and serious AEs in 17 (35 %). Grade 3 AEs were common with regorafenib treatment, which most frequently were skin reactions of the hand and foot, diarrhea, renal failure, fatigue, and hypertension. Two patients had grade 4 treatment-related AEs and 4 patients died during study treatment or within 30 days of their last dose.
14.2.5 Cabozantinib
MET/hepatocyte growth factor (HGF) is a signal involved in angiogenesis and malignant behavior [27]. It appears to be an important resistance pathway for VEGF-targeted therapy because MET levels increase after exposure to anti-angiogenic therapy [28]. Cabozantinib was identified as a dual inhibitor of VEGFR-2 and MET and shows additional activity against RET, KIT, AXL, and FLT3 [29].
Twenty-five patients with advanced RCC were enrolled in a Phase II trial for cabozantinib [30]. All patients had at least one line of prior therapy with systemic agents, 50 % had three or more lines of prior therapy, and 88 % of the patients had prior anti-VEGF therapies. Fourteen patients (56 %) were previously treated with sunitinib. The cabozantinib dose of 140 mg per day was administered orally that was previously determined as the MTD. Twenty-one of 25 patients were in the intermediate-risk group according to the Heng criteria [31], and 3 were in the poor-risk group. Of the 25 evaluated patients, 7 (28 %) achieved PR, 13 had SD (52 %), and 1 (4 %) showed disease progression. The median PFS was 14.7 months, but the median OS could not be calculated with a median follow up of 14.7 months (range: 11.2–21.8 months). Four patients had bone metastases, and 3 of these 4 patients showed a response. Pain was significantly and durably palliated in 2 patients reporting bone pain. The most common AEs were fatigue, diarrhea, hypophosphatemia, and hyponatremia.
Cabozantinib appeared to be effective in treating advanced RCC. Two Phase III trials have been conducted for patients with advanced RCC. One of the trials compares cabozantinib to sunitinib in previously untreated patients with advanced RCC ( ClinicalTrials.gov ., NCT01835158). The other (the METEOR trial) compares cabozantinib (60 mg/day) to everolimus in patients with RCC who previously received treatment with at least one VEGF-TKI ( ClinicalTrials.gov ., NCT01865747).
14.2.6 Tivantinib (ARQ 197)
Papillary RCC is the second most common subtype of RCC. Patients with hereditary papillary RCC have mutations in c-MET, which is also present in sporadic papillary RCC [32]. Therefore, MET-target therapy appears to be an attractive strategy for this disease. Tivantinib is a selective and non-ATP competitive inhibitor of c-MET that has shown a higher selectivity for c-MET compared with other kinase inhibitors [33]. In a Phase I trial including 10 patients with RCC, 6 patients (60 %) achieved SD, with 4 of these 6 patients (2 clear cell RCC, 2 unspecified renal cancer) maintaining treatment for more than 24 weeks [34]. The dose-limiting toxicities for tivantinib were neutropenia, thrombocytopenia, vomiting, and dehydration. Clinical trials for tivantinib in papillary RCC are currently being conducted ( ClinicalTrials.gov ., NCT01688973).
14.2.7 Foretinib (XL880)
Foretinib is an oral multikinase inhibitor that targets c-MET, RON, AXL, Tie-2, and VEGFRs. Foretinib potently inhibits c-MET and VEGFRs with a subnanomolar concentration [35]. In a Phase II study of foretinib that examined 74 patients with papillary RCC, the ORR was 13.5 %, and the median PFS was 9.3 months. The presence of a germ line MET mutation predicted the response to foretinib. Five of 10 patients (50 %) with the mutation responded to foretinib. In contrast, 10 of 57 patients (17.5 %) without the mutation responded. The most common AEs were fatigue, hypertension, gastrointestinal toxicities, and nonfatal pulmonary emboli. Foretinib demonstrated efficacy in patients with advanced papillary RCC. Therefore, patients with papillary RCC-associated germ line MET mutations are expected to respond to foretinib with relatively high rate.
14.3 PI3K/Akt/mTOR Pathway Inhibitors
Although cytokine therapies including interferon and interleukin-2 have shown limited efficacy, mTORC1-targeted therapies such as temsirolimus and everolimus have shown an improved PFS and OS in patients with advanced RCC. In a Phase III trial, temsirolimus demonstrated a better PFS and OS when compared to IFN-α (PFS: 5.5 vs. 3.1 months; OS: 10.9 vs. 7.3 months) [5]. Following a Phase III trial that demonstrated everolimus had an improved PFS compared with placebo (4.9 vs. 1.9 months) in patients with mRCC who had progressed with VEGF-TKI therapies, everolimus was approved as a treatment for advanced RCC [6]. Many patients with mRCC are first controlled with mTORC1 inhibitors; however, most of these patients who responded to mTORC1 inhibitors eventually demonstrate relapse. Resistance to mTORC1 inhibitors is considered to occur via pathways that can compensate or serve as alternatives for mTORC1 signaling, and this may lead to HIF accumulation [36]. Possible mechanisms (Fig. 14.1) for acquired mTORC1 inhibitor resistance are dysfunctional negative feedback loops that function normally during mTORC1/S6K pathway activity [37, 38]. One feedback loop prevents mTOR complex 2 (mTORC2) activation. Therefore, mTORC1/S6K inhibition leads to mTORC2-dependent AKT activation and HIF upregulation [37–41]. Another possible mechanism for mTORC1 inhibitor resistance is an inactive negative feedback loop that normally prevents activation of insulin receptor substrate 1 (IRS1)/PI3K/Akt signaling. Considering S6K typically suppresses IRS1 activation through the negative feedback loop, IRS1 activation occurs with mTORC1/S6K pathway inhibition. IRS1 activation subsequently activates HIF via the IRS1/PI3K/Akt pathway [37, 41–44]. Furthermore, HIF-2α is considered to have a greater contribution to RCC development and progression as compared with HIF-1α [46–48]. Recent studies demonstrated that HIF-2α translation requires mTORC2 activity [50], which indicates mTORC2 inhibition is a potentially viable treatment for RCC. The proposed mechanisms for mTORC1 therapy resistance have led to the development of agents targeting PI3K, Akt, and mTORC2.
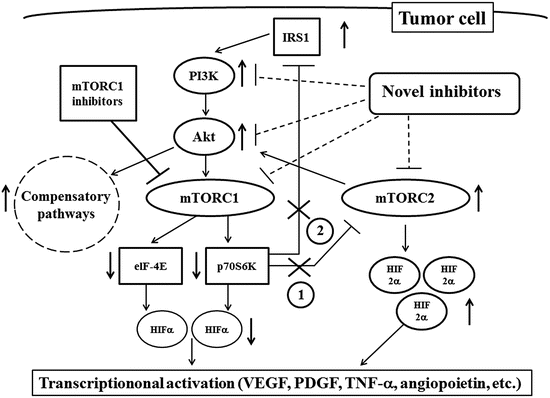
Fig. 14.1
Possible mechanisms for resistance of RCC cells to mTORC1 inhibitors. When mTORC1/S6 K pathway is active, one feedback loop prevents mTORC2 and Akt activation. Therefore, mTORC1/S6 K inhibition leads to mTORC2-dependent HIF upregulation and AKT activation (circle 1). Another feedback loop prevents activation of IRS1/PI3K/Akt signaling (circle 2) when mTORC1/S6 K pathway is active. mTORC1/S6 K inhibition leads to IRS1 activation and HIF upregulation through the IRS1/PI3K/Akt pathway. Novel inhibitors of PI3K/Akt/mTOR pathway which inhibit PI3K and/or Aki and/or mTORC2 activity have been developed
14.3.1 NVP-BEZ235
NVP-BEZ235 is a novel dual PI3K/mTORC1/2 inhibitor. BEZ235 inhibits class 1 PI3K activity by binding to its ATP-binding domain [49]. BEZ235 also binds directly to the mTOR ATP-binding domain and inhibits both TORC1 and TORC2. BEZ235 showed strong inhibition of cell proliferation; suppression of Akt, Mnk-1, eIF4E, and 4EBP-1 phosphorylation; and suppression of cyclin D1 and HIF-2α levels in RCC cell lines [50]. Furthermore, BEZ235 inhibited tumor growth in RCC tumor xenografts (from 786-O) with inhibitory effects resulting from its antiproliferative and pro-apoptotic activity [50]. In another experiment, RCC cell lines (786–0 and Caki-1) were treated with BEZ235 alone, sorafenib alone, and these two agents combined. The antitumor efficacy (reduced proliferation and increased apoptosis) of BEZ235 in combination with sorafenib was superior to NVP-BEZ235 or sorafenib alone [51]. In a multicenter, open-label Phase I trial in 59 patients with advanced solid tumors, including breast cancer and RCC, BEZ235 was well tolerated and demonstrated preliminary efficacy [52]. The common AEs were nausea, vomiting, diarrhea, fatigue, asthenia, anemia, and anorexia. A Phase I/II trial for BEZ235 was completed in patients with advanced RCC, the results of which will be revealed in the near future ( ClinicalTrials.gov ., NCT01453595). A Phase I trial for BEZ235 combined with everolimus is being tested in patients with advanced solid tumors including mRCC ( ClinicalTrials.gov ., NCT01482156 ).
14.3.2 GDC-0980
GDC-0980 is a selective and potent inhibitor of class I PI3K and mTORs (TORC1/2) [53]. In preclinical studies, GDC-0980 has shown activity against several cancer cell lines (RCC cell line was not included) and xenograft models [53, 54]. A Phase I study was conducted for advanced solid tumors that enrolled 33 patients. GDC-0980 treatment showed tumor shrinkage in 3 patients with mesothelioma (29–64 % decrease), a patient with a gastrointestinal stromal tumor (58 % decrease) who remained on study for more than 6 months, and a patient with adrenal cell carcinoma who remained on study for more than 1 year [55]. The most common AEs observed in more than 10 % of patients were fatigue, diarrhea, decreased appetite, nausea, rash, mucositis, hyperglycemia, vomiting, and constipation. A Phase II trial for GDC-0980 in comparison with everolimus in patients with mRCC who have progressed after a VEGF-targeted therapy is currently underway ( ClinicalTrials.gov ., NCT01442090 ).
14.3.3 Perifosine (KRX-0401)
Akt is a possible therapeutic target for RCC. Perifosine is a synthetic substituted heterocyclic alkylphosphocholine that can inhibit Akt activity [56]. Perifosine interfered with phosphoinositide metabolism, inhibition of protein kinase C, and inhibition of protein kinase B/Akt phosphorylation [57]. Perifosine targets the pleckstrin homology domain of Akt, preventing its translocation to the plasma membrane and subsequent activation by PDK1 and mTORC2 complexes. Perifosine has also been shown to block cell cycle progression by inducing p21 WAF1 [58, 59]. Two independent Phase II trials (Perifosine 228 and 231) have been conducted. In one Phase II trial (Perifosine 228 trial), 24 patients with advanced RCC received oral perifosine (100 mg daily) [60]. One patient achieved a PR (ORR = 4 %) and 11 patients (46 %) had a SD. The median PFS was 14.2 weeks. Another Phase II trial (Perifosine 231) includes two groups of patients that received perifosine (100 mg daily). Group A included 32 patients who had not previously received mTOR inhibitor, whereas Group B included 18 patients who had received one prior mTOR inhibitor. In Group A, 4 patients experienced a confirmed objective PR (ORR = 13 %), and 9 patients experienced SD. The median PFS for Group A was 14.1 weeks. In Group B, 1 patient experienced a confirmed objective PR (ORR = 6 %) with a duration of 23 months and 7 patients experienced SD. The median PFS for Group B was 14 weeks. The overall median PFS for both Group A and Group B was 14 weeks (95 % CI, 12.9–20.7 weeks). The ORR for the Perifosine 231 trial was 10 %. Overall, perifosine was well tolerated. The most common AEs included nausea, diarrhea, musculoskeletal pain, and fatigue.
Related posts:
 Predictive and Prognostic Markers in Metastatic Renal Cell Carcinoma
Mammalian Targets of Rapamycin Inhibitors: Temsirolimus and Everolimus
Predictive and Prognostic Markers in Metastatic Renal Cell Carcinoma
Mammalian Targets of Rapamycin Inhibitors: Temsirolimus and Everolimus
 Molecular Genetics of Renal Cell Carcinoma
Molecular Genetics of Renal Cell Carcinoma
 Optimization of Therapy by Pharmacokinetic–Pharmacodynamic Analyses
Optimization of Therapy by Pharmacokinetic–Pharmacodynamic Analyses
 Treatment Overview
Treatment Overview
 Hereditary Renal Cell Carcinoma
Hereditary Renal Cell Carcinoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


