Fig. 15.1
Metabolism of purine nucleotides
15.2 Forodesine
Forodesine [BCX-1777, immucillin-H, 7-[(2S,3S,4R,5R)-3,4-dihydroxy-5-hydroxymethyl-2-pyrrolidinyl]-1,5-dihydro-4H-pyrrolo-[3,2-d]-pyrimidin-4-one, monohydrochloride] is a transition-state analog that potently inhibits PNP enzyme activity (Fig. 15.2) [12–15]. Forodesine has shown anticancer effects against several types of cancer cells in preclinical studies and is now being investigated for its clinical efficacy against T-cell malignancies, including T-cell leukemia and cutaneous T-cell lymphoma [12, 16, 17].
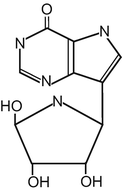
Fig. 15.2
The structure of forodesine
15.3 Mechanisms of Action
15.3.1 Mechanisms of Action of the Usual Nucleoside Analogs
The mechanisms of action of forodesine are different from those of the usual nucleoside analogs. After exposure to patients, these conventional nucleoside analogs are transported into cancer cells via nucleoside transporters and are then phosphorylated to their corresponding triphosphates. These analog triphosphates are subsequently incorporated into DNA by DNA polymerases, thereby inducing the termination of DNA synthesis and consequently inducing apoptosis [18–20]. In contrast to these nucleoside analogs, forodesine itself is not phosphorylated to its triphosphate form, nor is it incorporated into DNA strands.
15.3.2 Mechanisms of Action of Forodesine
In the blood, 2′-deoxyguanosine is converted to guanine and deoxyribose monophosphate via phosphorolysis in the presence of PNP. The guanine is then deaminated to xanthine, and the xanthine is subsequently metabolized to uric acid by xanthine oxidase (Fig. 15.3). If PNP is inhibited by forodesine, 2′-deoxyguanosine remains unmetabolized and is transported into cancer cells via nucleoside transporters. Inside the cells, 2′-deoxyguanosine is phosphorylated to deoxyguanosine monophosphate by deoxycytidine kinase and then to deoxyguanosine triphosphate by other kinases. The intracellular deoxyguanosine triphosphate inhibits ribonucleotide reductase, induces deoxyribonucleotide pool imbalance, and inhibits DNA synthesis and repair [15, 21–23]. Forodesine selectively inhibits the in vitro growth of malignant T-cell lines in the presence of 2′-deoxyguanosine. It is preferentially cytotoxic to T cells, because T cells possess a relatively high level of kinase activity and a relatively low level of nucleotidase activity [8].
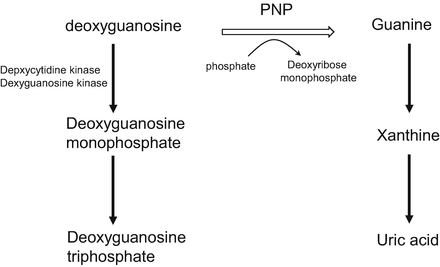
Fig. 15.3
Inhibition of purine nucleoside phosphorylase. PNP purine nucleoside phosphorylase
15.3.3 The Role of p53 in the Cytotoxic Activity of Forodesine
Balakrishnan et al. reported that when chronic lymphocytic leukemia lymphocytes were treated with forodesine and 2′-deoxyguanosine, intracellular deoxyguanosine triphosphate accumulated in the cells. This accumulation was associated with DNA damage-induced p53 stabilization, phosphorylation of p53 at Ser15, and activation of p21 [24]. In another study, Alonso et al. demonstrated that forodesine was highly cytotoxic as a single agent or in combination with bendamustine and rituximab in primary chronic lymphocytic leukemia cells. This cytotoxicity was independent of CD38/ZAP-70 expression and p53 or ATM deletion [25].
15.3.4 The Cytotoxicity of Forodesine in Combination with Nelarabine
We investigated the cytotoxicity of forodesine in combination with nelarabine in vitro using the human T-lymphoblastic leukemia cell line CCRF-CEM, because both agents are cytotoxic to T-cell malignancies [26]. Nelarabine is a purine nucleoside analog with anticancer properties that is used for treating refractory or relapsed T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. When the CCRF-CEM cells were treated with forodesine in the presence of 2′-deoxyguanosine, the forodesine was active and inhibited cell growth. However, the addition of 9-β-D-arabinofuranosylguanine, an active compound of nelarabine, to the forodesine and 2′-deoxyguanosine did not enhance the growth inhibition effect of forodesine. The combination index revealed antagonism between forodesine and 9-β-D-arabinofuranosylguanine. The intracellular production of 9-β-D-arabinofuranosylguanine triphosphate was reduced in the presence of forodesine. A CEM subclone resistant to 9-β-D-arabinofuranosylguanine was cross-resistant to forodesine. Thus, the combination of forodesine and nelarabine would not be an effective regimen for treating patients with T-cell malignancies.
15.4 Pharmacokinetics
15.4.1 Phase I Studies
A phase I clinical trial was conducted by Gandhi et al. in five patients with previously treated T-cell malignancies, such as T-cell lymphoblastic lymphoma, T-cell acute lymphoblastic leukemia, and T-cell prolymphocytic leukemia, as the first study of forodesine in humans [11]. Forodesine (40 mg/m2) was infused over 30 min on the first day. Plasma and cellular pharmacokinetics and pharmacodynamics were investigated. The median plasma peak level of forodesine (5.4 μM; range, 4.9–7.8 μM) occurred at the end of the infusion. The half-life of plasma forodesine was between 6 and 17 h. The plasma 2′-deoxyguanosine concentration after eight intravenous infusions of forodesine reached a maximum of 20 μM (range, 2.6–36.8 μM). The intracellular deoxyguanosine triphosphate concentrations were also determined in leukemic cells. After the administration of forodesine on day 1, deoxyguanosine triphosphate concentrations increased by five- to tenfold within the first 8 h and by 10- to 20-fold within the first 24 h after the start of therapy [11].
A phase I study was conducted in 13 Japanese patients with relapsed or refractory peripheral T/natural killer-cell malignancies [27]. Forodesine was administered orally once daily at 100, 200, or 300 mg. The mean maximum concentrations of plasma forodesine were 139.2 ng/mL (day 1) and 216.5 ng/mL (day 5) at 100 mg, 335.3 ng/mL (day 1) and 499.0 ng/mL (day 5) at 200 mg, and 328.0 ng/mL (day 1) and 421.6 ng/mL (day 5) at 300 mg. The area under the plasma concentration-time curve values at the doses of 100, 200, and 300 mg were 1948 ng·h/mL (day 1) and 2730 ng·h/mL (day 5), 4608 ng·h/mL (day 1) and 6303 ng·h/mL (day 5), and 4596 ng·h/mL (day 1) and 5587 ng·h/mL (day 5), respectively. As for the pharmacodynamics, the mean maximum concentrations of plasma 2′-deoxyguanosine were 270.4 ng/mL (day 1) and 315.5 ng/mL (day 15) at 100 mg, 348.7 ng/mL (day 1) and 410.7 ng/mL (day 15) at 200 mg, and 374.4 ng/mL (day 1) and 515.8 ng/mL (day 15) at 300 mg. The area under the plasma concentration-time curve values of 2′-deoxyguanosine at the doses of 100, 200, and 300 mg were 4023 ng·h/mL (day 1) and 5384 ng·h/mL (day 15), 5705 ng·h/mL (day 1) and 7696 ng·h/mL (day 15), and 6074 ng·h/mL (day 1) and 9533 ng·h/mL (day 15), respectively.
15.4.2 A Phase II Study
In a phase II study by Balakrishnan et al., 200 mg of forodesine was administered orally in eight patients with chronic lymphocytic leukemia [28]. On day 1, a plasma forodesine level of 300 nM was achieved (range, 126–600 nM). Overall, the plasma forodesine levels on days 2, 3, 4, and 5 ranged between 200 and 1300 nM. The level of 2′-deoxyguanosine in pretreated samples was below the level of detection. On day 1, the plasma concentration of 2′-deoxyguanosine reached a median of 1.2 μM (range, 0.57–2.2 μM), and on day 27, it was between 1.1 and 2.3 μM.
15.5 The Method of Administration
Clinical studies of forodesine are underway. In such studies, forodesine is mainly administered orally once a day at 200–600 mg. The bioavailability of forodesine after oral administration was 40–59% in humans [29].
15.6 Toxicities
15.6.1 Phase I Studies
In the phase I study performed by Gandhi et al., the most common toxicity observed was grade 3–4 neutropenia associated with infection. Other grade 3–4 toxicities included dyspnea, renal failure, depression, and hypoxemia. The grade 1–2 toxicities observed during the period of forodesine infusion were hyperbilirubinemia, hypotension, headache, hypocalcemia, tremors, constipation, and nausea/vomiting [11]. Hematologic toxicities other than neutropenia were inevaluable owing to baseline cytopenias.
In the Japanese phase I study, the grade 3–4 adverse events included lymphocytopenia, anemia, leukocytopenia, and pyrexia. Laboratory test results showed no remarkable changes, and vital signs and body weight showed no consistent changes during the study treatment period. None of the patients showed any new, clinically significant abnormal findings on 12-lead electrocardiograms, and none of them developed any dose-limiting toxicities due to the treatment with forodesine at the designated dose range of 100–300 mg [27].
15.6.2 Phase II Studies
The phase II trial reported by Balakrishnan et al. demonstrated mild adverse events in patients with chronic lymphocytic leukemia treated with oral forodesine [28]. The adverse events were fatigue, bronchitis/pneumonia, diarrhea, fever, neutropenia, and thrombocytopenia.
In the phase II study by Dummer et al., the most commonly reported adverse events were peripheral edema, fatigue, insomnia, pruritus, diarrhea, headache, and nausea [30]. Severe adverse events included anemia, lymphocytopenia, arterial fibrillation, fatigue, edema, Herpes zoster, skin infection, CD4 lymphocyte depletion, depression, pruritus, and rash.
Apart from these reports, serum uric acid level decreases have also been reported as a consequence of PNP inhibition (Fig. 15.1).
15.7 Clinical Studies (Table 15.1)
15.7.1 Phase I Studies
The first-ever clinical study of forodesine was performed by Gandhi et al. [11]. Five patients (three T-cell acute lymphoblastic leukemias, two T-cell prolymphocytic leukemias) were treated with an intravenous infusion of forodesine (40 mg/m2) on day 1; treatment continued on day 2, and thereafter, forodesine was administered every 12 h for an additional eight doses. The dose of forodesine was increased to 90 mg in the subsequent courses in one patient. Overall, no objective responses were observed. Three patients had stable disease after one course of forodesine. The disease progressed in two patients.
Table 15.1




Clinical studies of forodesine in patients with hematological malignancies
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
