Fig. 1
×200 image of a 4-mm-thick human tonsil section placed directly into 40 °C 10 % neutral buffered formalin for 1 h. Sample is stained with hematoxylin and eosin to reveal tissue morphology
In order to make a broadly adaptable fixation protocol it was first necessary to think about the chemical principles that make formaldehyde a good fixative. Since fixation is a two-step process driven by diffusion and crosslinking, we focused on the optimization of these two major chemical functions. Diffusion is largely driven by concentration gradient. This scenario exists when the tissue is first placed in the fixative, where there is an overwhelming excess of NBF on the outside and none in the middle. As time passes, a gradient forms where formalin penetrates the tissue, until sufficient amounts of formaldehyde have accumulated to drive the crosslinking reaction. The diffusion of formaldehyde can be accomplished under a wide range of temperatures as this molecule is very small. However, the second step of crosslinking is largely driven by concentration and temperature. As in any other chemical reaction, crosslinking needs a minimal concentration of reactant (formaldehyde) to drive the reaction to completion (crosslinked biomolecule). Increasing the temperature shifts the chemical reaction to favor crosslinking and if sufficient concentrations of formaldehyde are present fixation happens rapidly. In order to alleviate the fixation gradient that forms with heated reactions, we reasoned that it would be necessary to allow sufficient diffusion of formaldehyde before increasing the temperature to initiate crosslinking.
This was accomplished by employing a two-temperature fixation protocol with standard 10 % NBF (Fig. 2). In this scenario, tissues are placed in cold formalin (optimally 4 °C). Under these conditions, little crosslinking occurs (data not shown) but diffusion still proceeds. By cooling the tissues, cellular enzymes are largely inactivated and the cold environment helps preserve biomolecules, especially posttranslational modifications. After a period of cool time, depending on tissue thickness, samples are placed directly into warm formalin (optimally 45 °C) favoring formation of crosslinks. Since formalin is already present throughout the tissue sample, crosslinking occurs evenly throughout the tissue and superior cellular structure is realized (Fig. 3, H&E). In addition, biomolecules are better preserved due to the almost immediate decrease of cellular enzyme activity (Fig. 3, IHC/bcl-2). For tissues up to 4 mm in thickness, 2 h of cold followed by 2 h of warm formalin fixation works best (Fig. 4b, c, see bold boxes). We therefore named this the “2 + 2” fixation protocol.
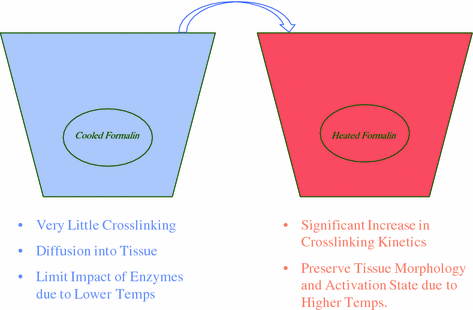
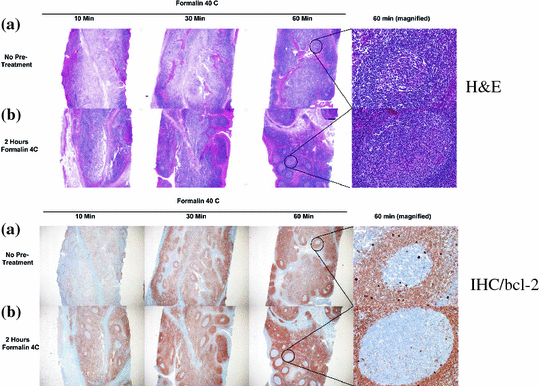

Fig. 2
Sequence of events to carry out cold + warm tissue fixation. Tissue is first placed into cooled formalin at 4 °C for 2 h followed by immediate transfer and immersion into formalin heated at 45 °C for 2 h
Fig. 3
Top two panels show a time course of formalin fixation of 4-mm-thick human tonsil sample with hematoxylin and eosin stains. a Warm only formalin fixation, b cold + warm formalin fixation. Right side panels show ×400 magnification of center germinal centers. Bottom two panels show the same set of samples stained with an antibody that recognizes bcl-2 protein and developed with DAB (Brown) on a Ventana Medical Systems Discovery XT Automated Staining platform
Fig. 4
Heat maps of tissue fixation quality. Left panel shows 4-mm-thick human tonsil samples fixed with room temperature 10 % NBF (Control). Tissues were immersed in 10 % NBF for indicated hours (0–24 h). Colors indicate quality of resulting cellular morphology. Green excellent, Yellow Good, Red poor. For all samples, tissues were deemed excellent after 8 h in RT formalin (Green shading). Middle panel shows hematoxylin and eosin stains of 4 mm human tonsil samples placed into heated formalin after a cold soak pretreatment (2 h in 4 °C 10 % NBF). Samples were then placed into 10 % NBF at indicated temperatures (Gray shaded boxes) for indicated times (0–6 h). 4 mm human tonsil samples placed into heated formalin after a cold soak pretreatment (2 h in 4 °C 10 % NBF). Samples were then placed into 10 % NBF at indicated temperatures (Gray shaded boxes) for indicated times (0–6 h). Bold black boxes indicate the area of the heat map for the 2 + 2 method
The use of heat as a means of decreasing fixation time is proven to be useful and can alleviate some of the workflow bottlenecks in standard histology operations. However, the use of excessive heat was detrimental to downstream assays that depend on epitope recognition. After a 2 h incubation in cold formalin, crosslinking in temperatures higher than 45 °C resulted in a severe decrease in immunohistochemical signaling (Fig. 4c, see 2–6 h at 50 °C). Since the fixation of tissues needs to be universal for all downstream assays, 2 h at 45 °C provides the maximum staining for both tissue morphology (H&E) as well as antigen recognition (IHC) and nucleic acid-based assays (Fig. 4 and data not shown).
3 Clinical Validation
The 2 + 2 fixation method was tested under clinical conditions to assess if this protocol would be truly useful for clinical practices. This study utilized tissue collection of unfixed surgical samples of >20 tissue types that was considered waste material in a clinical laboratory setting [10]. Surgical tissues were fixed with the 2 + 2 protocol, and conventionally fixed tissue with increasing times of room temperature 10 % NBF (0, 2, 4 and 24 h) as comparison. Hematoxylin and eosin stains were performed on both sets of tissue to examine cellular morphology. Slides were scored by a board-certified pathologist by blinding the samples and examining nuclear, cytoplasmic, and general morphology of the tissues. Each category was given a score up to 2 and added together to make up a “histologic score,” a total of 6 indicating excellence. A summary of all of the data points in the experiment are shown in Fig. 5 where increasing numbers on the Y-axis represent better morphology. As can be seen, samples that received no fixation on average score very poorly, with many scores at or below 2. However, as fixation time is increased, data points migrate up the graph to reveal better morphology until at 24 h, most of the samples have histologic scores of 5–6. In comparison, the 2 + 2 method compared favorably with the 24 h fixation times and with a gold standard sample that consisted of a stain of the actual clinical sample used for diagnosis. These samples were also compared in several antibody and nucleic acid-based assays. In each case, the 2 + 2 method compared favorably with 24 h standard fixed samples (data not shown).
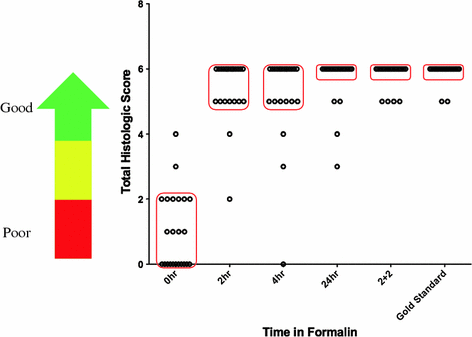
Fig. 5
Summary of all samples comparing the 2 + 2 protocol with standard room temperature fixation protocol. Over 30 different tissue types were fixed and stained with hematoxylin and eosin and scored for morphological details. 0–2 points were given for nuclear detail, cytoplasmic detail and general morphology. A score of 6 signals excellence, see calibration arrow at left of graph. Samples were fixed either with 2 + 2 or 0, 2, 4, and 24 h with room temperature formalin as indicated. All fixation methods were compared to a gold standard sample consisting of a cut and stain of the actual clinical specimen used for pathological review
4 Phosphomarker Preservation and Personalized Medicine
Many cancer pathways utilize a variety of protein activation cascades to carry growth signals from the outside of the cell to internal cellular compartments. While there are many types of activation signals, the most prominent is the addition of phosphate groups to serine, threonine, or tyrosine positions on the protein backbone. Addition of phosphate groups alters the tertiary structure of the protein target and often times “activates” an enzymatic activity that is normally masked. This new activity can then act on the next protein target in the pathway until the last protein in the pathway acts on a cellular function causing either an increase in cellular growth, lack of cell death, or some other vital function. The addition of phosphates is carried out by kinases, and their removal by enzymes called phosphatases. These two enzymes are a tightly regulated pair used for the control of cell growth and regulation of cellular functions such as DNA replication, RNA transcription, and protein translation. Study of these pathways of activation is of high importance and gaining traction in the age of personalized medicine [16–21]. Many proteins that were once unavailable as new drug candidates are becoming targets for the next generation of pharmaceuticals. Hindering these efforts are the unstandardized fixation methods that simply cannot preserve activation states at levels necessary for assay development. One such marker, pAKT (also known as protein kinase B (PKB)), resides in the heavily studied PI3 K pathway, but researchers have not been able to fully preserve the protein in its activated state [22–24]. This molecule is phosphorylated at the cellular membrane and migrates into the cytoplasm where it acts on proteins in various signaling pathways. Due to the transient nature of the activated form, no reliable assays utilizing this important biomarker have been developed for tissue diagnostics.
Related posts:
 Quality in Fresh-Frozen Gastrointestinal Tumor Specimens—Experiences from the Tumor and Healthy Tissue Bank TU Dresden
Quality in Fresh-Frozen Gastrointestinal Tumor Specimens—Experiences from the Tumor and Healthy Tissue Bank TU Dresden
 in Lung Cancer
in Lung Cancer
 Pre-analytical Phase in Surgical Pathology
Pre-analytical Phase in Surgical Pathology
 Formalin Fixation and Rapid Microwave Processing Do Not Affect HER2 Testing
Formalin Fixation and Rapid Microwave Processing Do Not Affect HER2 Testing
 the Lean lab: The Industry Challenge
the Lean lab: The Industry Challenge
 Validation of Vacuum Sealed, Formalin-Free Tissue Preservation, and Transport System
Validation of Vacuum Sealed, Formalin-Free Tissue Preservation, and Transport System
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


