SUMMARY
Improved understanding of the molecular mechanisms of fibrinolysis has led to major advances in fibrinolytic and antifibrinolytic therapy. Characterization of the genes for all the major fibrinolytic proteins has revealed the structure of the relevant serine proteases, their inhibitors, and their receptors. The development of genetically engineered animals deficient in one or more fibrinolytic protein(s) has revealed both expected and unexpected functions. In addition, we now have a catalog of acquired and inherited disorders reflective of either fibrinolytic deficiency with thrombosis or fibrinolytic excess with hemorrhage. These advances have led to development of more effective and safer protocols for both pro- and antifibrinolytic therapy in a variety of circumstances.
In response to vascular injury, fibrin, the insoluble end product of the action of thrombin on fibrinogen, is deposited in blood vessels, thus stemming the flow of blood. Once the vessel has healed, the fibrinolytic system is activated, converting fibrin to its soluble degradation products through the action of the serine protease, plasmin (Fig. 25–1A). Fibrinolysis is subject to precise control because of the actions of multiple activators, inhibitors, and cofactors.1 In addition, receptors expressed by endothelial, monocytoid, and myeloid cells provide specialized, protected environments where plasmin can be generated without compromise by circulating inhibitors (Fig. 25–1B).2,3 Beyond its more traditional role in fibrin degradation, the fibrinolytic system also supports a variety of tissue remodeling mechanisms. This chapter reviews the fundamental features of plasmin generation, considers the major clinical syndromes resulting from abnormalities in fibrinolysis, and discusses approaches to fibrinolytic and antifibrinolytic therapy.
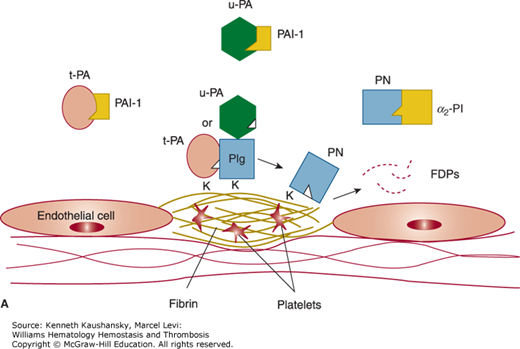
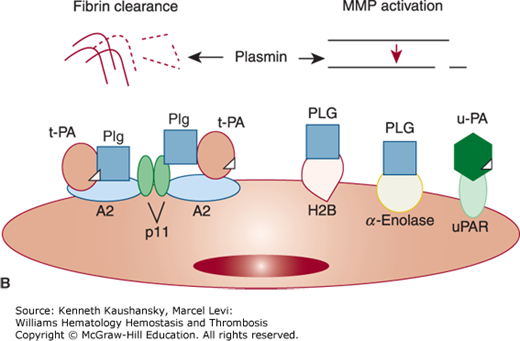
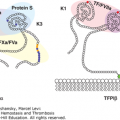
Figure 25–1.
Overview of the fibrinolytic system. A. Fibrin-based plasminogen activation. The zymogen plasminogen (Plg) is converted to the active serine protease, plasmin (PN), through the action of tissue-type plasminogen activator (t-PA) or urokinase-type plasminogen activator (u-PA). The activity of t-PA is greatly enhanced by its assembly with Plg through lysine residues (K) on a fibrin-containing thrombus. u-PA acts independently of fibrin. Both t-PA and u-PA can be inhibited by plasminogen activator inhibitor-1 (PAI-1), the main physiologic regulator of plasminogen activator activity. By binding to fibrin, PN is protected from its major inhibitor, α2-plasmin inhibitor (α2–PI). Fibrin-bound plasmin degrades crosslinked fibrin, giving rise to soluble fibrin degradation products (FDPs). B. Cell surface plasminogen activation. Although many cell types express receptors for Plg, urokinase, and t-PA, only the endothelial cell is depicted here. The annexin A2 heterotetramer, consisting of two copies each of annexin A2 (A2) and protein p11 (p11), binds both t-PA and Plg, thereby augmenting the efficiency of plasmin generation on endothelial cells. Plg may also bind to other endothelial cell receptors, including histone H2B (H2B) and α-enolase, and may be activated by u-PA bound to its receptor, uPAR, to effect plasmin generation.
Synthesized primarily in the liver,4,5 plasminogen is a Mr approximately 92,000 single-chain proenzyme that circulates in plasma at a concentration of approximately 1.5 μM6 (Table 25–1). The plasma half-life of plasminogen in adults is approximately 2 days.7 Its 791 amino acids are crosslinked by 24 disulfide bridges, 16 of which give rise to five homologous triple loop structures called “kringles” (Fig. 25–2).8 The first (K1) and fourth (K4) of these 80-amino-acid, Mr approximately 10,000 structures impart high- and low-affinity lysine binding, respectively.9 The lysine-binding domains of plasminogen appear to mediate its specific interactions with fibrin, cell surface receptors, and other proteins, including its circulating inhibitor α2-plasmin inhibitor (α2-PI).10–14
| A. Major Proteases | |||||
| Property | Plasminogen | t-PA | u-PA | ||
| Molecular mass | 92,000 | 72,000 | 54,000 | ||
| Amino acids | 791 | 527 | 411 | ||
| Chromosome | 6 | 8 | 10 | ||
| Site of synthesis | Liver | Endothelium | Endothelium, kidney | ||
| Plasma concentration | |||||
| nM | 1500 | 0.075 | 0.150 | ||
| mcg/mL | 140 | 0.005 | 0.008 | ||
| Plasma half-life | 48 h | 5 min | 8 min | ||
| N-Glycosylation (%) | 2 | 13 | 7 | ||
| Form 1 | – | Asn117, Asn184, Asn448 | Asn302 | ||
| Form 2 | Asn288 | Asn117, –, Asn448 | – | ||
| O-Glycosylation | |||||
| α-Fucose | – | Thr61 | Thr18 | ||
| Complex | Thr345 | – | – | ||
| Two-chain cleavage site | Arg560-Val561 | Arg275-Ile276 | Lys158-Ile159 | ||
| Heavy-chain domains | |||||
| Finger | No | Yes | No | ||
| Growth factor | No | Yes | Yes | ||
| Kringles (no.) | 5 | 2 | 1 | ||
| Light-chain catalytic triad | His602, Asp645, Ser740 | His322, Asp371, Ser478 | His204, Asp255, Ser356 | ||
| B. Major Serpin Inhibitors | |||||
| Property | α2-PI | PAI-1 | PAI-2 | ||
| Molecular mass | 70,000 | 52,000 | 60,000 (glycosylated) 47,000 (nonglycosylated) | ||
| Amino acids | 452 | 402 | 393 | ||
| Chromosome | 18 | 7 | 18 | ||
| Sites of synthesis | Kidney, liver | Endothelium | Placenta | ||
| Monocytes/macrophages | Monocytes/macrophages | ||||
| Hepatocytes | Tumor cells | ||||
| Adipocytes | |||||
| Plasma concentration | |||||
| nM | 900 | 0.1–0.4 | ND | ||
| mcg/mL | 50 | 0.02 | ND | ||
| Serpin reactive site | Arg364–Met365 | Arg346–Met347 | Arg358–Thr359 | ||
| Specificity | Plasmin | u-PA = t-PA | u-PA > t-PA | ||
| C. Major Activation Receptors | |||||
| Property | uPAR | Annexin A2 | p11 | Plg-RKT | Histone 2B |
| Molecular mass | 55,000–60,000 | 36,000 | 11,000 | 17,000 | 17,000 |
| Amino acids | 313 | 339 | 4544 | 147 | 126 |
| Chromosome | 19 | 15 | 12 | 9 | 6 |
| Source | Endothelial cells | Endothelial cells | Endothelial cells | – | Endothelial cells |
| Monocytes | Monocytes | Monocytes | Monocytes | Monocytes | |
| Macrophages | Macrophages | Macrophages | Macrophages | Macrophages | |
| Fibroblasts | Myeloid cells | Myeloid cells | Myeloid cells | Neutrophils | |
| Tumor cells | Tumor cells | Tumor cells | Tumor cells | ||
| Ligand(s) | u-PA | t-PA, Plg | Plg | Plg | |
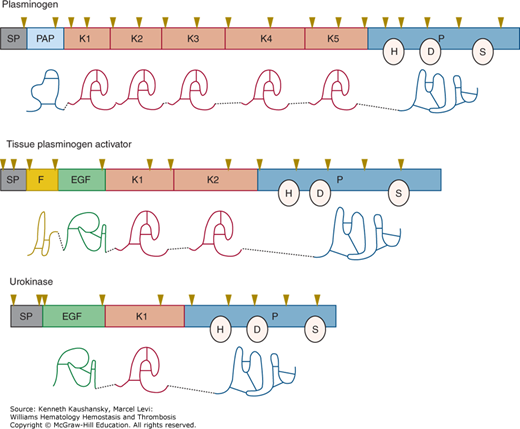
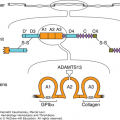
Figure 25–2.
Alignment of the intron–exon structure of plasminogen, tissue plasminogen activator, and urokinase genes showing functional protein domains. Protein domains are labeled signal peptide (SP), preactivation peptide (PAP), “kringle” domains (K), fibronectin-like “finger” (F), epidermal growth factor–like domain (EGF), and protease (P). The positions of the catalytic triad amino acids histidine (H), aspartic acid (D), and serine (S) are shown within individual protease domains. The positions of individual introns relative to amino acid encoding exons are indicated with inverted triangles.
Acronyms and Abbreviations:
α2-PI, alpha-2 plasmin inhibitor; APL, acute promyelocytic leukemia; CI, confidence interval; CT, computed tomography; DIC, disseminated intravascular coagulation; HC, homocysteine; IL, interleukin; LDL, low-density lipoprotein; MMP, matrix metalloproteinase; MRI, magnetic resonance imaging; Plg, plasminogen; PAI, plasminogen activator inhibitor; TAFI, thrombin-activatable fibrinolysis inhibitor; TGF-β, transforming growth factor beta; t-PA, tissue-type plasminogen activator; u-PA, urokinase-type plasminogen activator; uPAR, urokinase-type plasminogen activator receptor.
Posttranslational modification of plasminogen results in two glycosylation variants (forms 1 and 2; see Table 25–1).15–17 O-linked oligosaccharide, consisting of sialic acid, galactose, and galactosamine resident on Thr345, is common to both forms. Only form 2, however, contains N-linked oligosaccharide on Asn 288 that is comprised of sialic acid, galactose, glucosamine, and mannose. The carbohydrate portion of plasminogen appears to regulate its affinity for cellular receptors and may also specify its physiologic degradation pathway.
Activation of plasminogen results from cleavage of a single Arg–Val peptide bond at position 560–561,6 giving rise to the active protease, plasmin (see Table 25–1). Plasmin contains a typical serine protease catalytic triad (His 602, Asp 645, and Ser 740), but exhibits broad substrate specificity when compared to other proteases of this class.18 The circulating form of plasminogen, aminoterminal glutamic acid plasminogen (Glu–Plg), can be converted by limited proteolysis to several modified forms known collectively as Lys–Plg.19,20 Hydrolysis of the Lys77–Lys78 peptide bond gives rise to a conformationally modified form of the zymogen that more readily binds fibrin, displays two- to threefold higher avidity for cellular receptors, and is activated 10 to 20 times more rapidly than Glu–Plg.11,21,22 Lys–Plg does not normally circulate in plasma21 but has been identified on cell surfaces.23,24
Spanning 52.5 kb of DNA on chromosome 6q26–27, the Plg gene consists of 19 exons25,26 and directs expression of a 2.7-kb mRNA8 (see Fig. 25–2). The 5′ upstream region of the Plg gene contains two regulatory elements common to genes for acute-phase reactants (CTGGGA) and six interleukin (IL)-6 response elements.26 Plg gene activity, moreover, is stimulated by the acute-phase-mediator IL-6 both in vitro and in vivo.27 The gene is closely linked and structurally related to that of apolipoprotein(a), an apoprotein associated with the highly atherogenic low-density lipoprotein–like particle lipoprotein(a),28 and more distantly related to other kringle-containing proteins such as tissue-type plasminogen activator (t-PA), urokinase-type plasminogen activator (u-PA), macrophage-stimulating protein, and hepatocyte growth factor.29–34
Mice made completely deficient in Plg through gene targeting undergo normal embryogenesis and development, are fertile, and survive to adulthood (Table 25–2).35,36 These animals display runting and ligneous conjunctivitis,37 and harbor spontaneous thrombi in the liver, stomach, colon, rectum, lung, and pancreas, as well as fibrin deposition in the liver and ulcerative lesions in the gastrointestinal tract and rectum. These results suggested that Plg is not strictly required for normal development, but does play a central role in fibrin homeostasis. In humans, Plg deficiency presents most often with ligneous mucositis as a result of fibrin deposition and is rarely a cause of macrovascular thrombosis (see “Fibrinolytic Deficiency and Thrombosis” below).
| Genotype | Some Phenotypic Features | References |
|---|---|---|
| Plasminogen | ||
| Plg–/– | Spontaneous thrombosis, runting, premature death | 35, 36 |
| Fibrin in liver, lungs, stomach; gastric ulcers | 35, 36 | |
| Impaired wound healing | 243, 244 | |
| Ligneous mucositis | 37 | |
| Impaired monocyte recruitment | 245 | |
| Impaired neointima formation after electrical injury | 246 | |
| Impaired dissemination of Borrelia burgdorferi | 247 | |
| Plasminogen Activators | ||
| t-PA–/– | Reduced lysis of fibrin clot | 84 |
| Increased endotoxin-induced thrombosis | 84 | |
| u-PA–/– | Occasional fibrin in liver/intestine | 84 |
| Rectal prolapse, ulcers of eyelids, face, ears | 84 | |
| Reduced macrophage degradation of fibrin | 84 | |
| Increased endotoxin-induced thrombosis | 84 | |
| u-PA–/– t-PA–/– | Reduced growth, fertility, and life span; cachexia | 84 |
| Fibrin deposits in liver, gonads, lungs | 84 | |
| Ulcers in intestine, skin, ears; rectal prolapse | 84 | |
| Impaired clot lysis | 84 | |
| Inhibitors | ||
| α2PI–/– | Reduced fibrin deposition following endotoxin | 90 |
| Enhanced lysis of injected plasma clots | 90 | |
| PAI-1–/– | Mildly increased lysis of fibrin clot | 123 |
| Resistance to endotoxin-induced thrombosis | 124 | |
| TAFI–/– | Increased clot lysis | 140, 142 |
| Reduced injury-related venous thrombosis | 141 | |
| Receptors | ||
| uPAR–/– | Normal development and fertility | 163 |
| Normal clot lysis | 164 | |
| Annexin A2–/– | Fibrin deposition in microvasculature | 205 |
| Impaired clearance of arterial thrombi | 205 | |
| Impaired postnatal neoangiogenesis | 198, 205, 270 | |
| S100A10–/– | Reduced baseline fibrin deposition | 199 |
One of two major endogenous Plg activators, t-PA consists of 527 amino acids comprising a glycoprotein of Mr approximately 72,000 (see Table 25–1).38 t-PA contains five structural domains including a fibronectin-like “finger,” an epidermal growth factor–like domain, two “kringle” structures homologous to those of Plg, and a serine protease domain (see Fig. 25–2). Cleavage of the Arg275–Ile276 peptide bond by plasmin converts t-PA to a disulfide-linked, two-chain form.38 Although single-chain t-PA is less active than two-chain t-PA in the fluid phase, both forms demonstrate equivalent activity when fibrin bound.39
The two glycosylation forms of t-PA are distinguishable by the presence (type 1) or absence (type 2) of a complex N-linked oligosaccharide moiety on Asn184 (see Table 25–1).40,41 Both types, however, contain high mannose carbohydrate on Asn 117, complex oligosaccharide on Asn448, and an O-linked α-fucose residue on Thr61.42 The carbohydrate moieties of t-PA may modulate its functional activity, regulate its binding to cell surface receptors, and specify its degradation pathways.
Located on chromosome 8p12–q11.2, the gene for human t-PA is encoded by 14 exons spanning a total of 36.6 kb (see Fig. 25–2).43–45 Although exon 1 encodes a 58-nucleotide mRNA leader sequence, each of the structural domains of t-PA is encoded by one or two of the remaining 13 exons. This arrangement suggests that the t-PA gene arose by an evolutionary process called “exon shuffling,” whereby functionally related genes evolved through rearrangement of exons encoding autonomous domains. Consistent with this hypothesis, deletion of exons encoding the fibronectin-like finger or kringle 2, but not kringle 1, domains of t-PA results in expression of mutants resistant to the cofactor activity of fibrin, while catalytic activity in the absence of fibrin remains intact.46
The proximal promoter of the human t-PA gene contains binding sequences for potentially important transcriptional factors including AP1, NF1, SP1, and AP2,47,48 as well as a potential cyclic adenosine monophosphate (cAMP)–responsive element (CRE).49 In vitro, many agents have been shown to exert small effects on the expression of t-PA mRNA, but relatively few enhance t-PA synthesis without augmenting plasminogen activator inhibitor (PAI)-1 synthesis as well. Agents that regulate t-PA gene expression independently of PAI-1 include histamine, butyrate, retinoids, arterial levels of shear stress, and dexamethasone.50–55 Forskolin, which increases intracellular cAMP levels, has been reported to decrease synthesis of both t-PA and PAI-1.48,56
In the vascular system, t-PA is synthesized and secreted primarily by endothelial cells belonging to a restricted set of blood vessels. In rodents, t-PA expression appears in 7- to 30-μm diameter precapillary arterioles in the lung, postcapillary venules, and vasa vasorum; much less expression is seen in endothelial cells of the femoral artery, femoral vein, carotid artery, or aorta.57 In the mouse lung, bronchial arteriolar endothelial cells express t-PA antigen, especially at branch points, while pulmonary blood vessels are uniformly negative.51,58–60 t-PA has also been detected in sympathetic neurons associated with the blood vessel wall.61 Release of t-PA is governed by a variety of stimuli such as thrombin, histamine, bradykinin, epinephrine, acetylcholine, arginine vasopressin, gonadotropins, exercise, venous occlusion, and shear stress.50,51,62,63 Its circulating half-life is exceedingly short (~5 minutes). Alone, t-PA is actually a poor activator of Plg, but, in the presence of fibrin, the catalytic efficiency of t-PA–dependent plasmin generation (kcat/Km) increases by at least two orders of magnitude.22 This is the result of a dramatic increase in affinity (decreased Km) between t-PA and its substrate Plg in the presence of fibrin. Although it is also expressed by extravascular cells, t-PA appears to represent the major intravascular activator of Plg.18
The second endogenous Plg activator, single-chain u-PA or prourokinase, is an Mr approximately 54,000 glycoprotein consisting of 411 amino acids (see Table 25–1). u-PA possesses an epidermal growth factor–like domain, a single Plg-like “kringle,” and a classical catalytic triad (His204, Asp255, Ser356) within its serine protease domain (see Fig. 25–2).64 Cleavage of the Lys158–Ile159 peptide bond by plasmin or kallikrein converts single-chain u-PA to a disulfide-linked two-chain derivative.65 Located on chromosome 10, the human u-PA gene is encoded by 11 exons spanning 6.4 kb and expressed by activated endothelial cells, macrophages, renal epithelial cells, and some tumor cells.66,67 Its intron–exon structure is closely related to that of the t-PA gene. u-PA expression appears to be induced during neoplastic transformation, possibly through the action of transcription factors AP1 and AP2.68 Other in vitro u-PA inducers include hormones, angiogenic growth factors, and cAMP,55 as well as tumor necrosis factor and transforming growth factor-β (TGF-β).69–71
Two-chain u-PA occurs in both high- (Mr 54,000) and low-molecular-weight (Mr 33,000) forms that differ by the presence or absence, respectively, of a 135-residue aminoterminal fragment released by plasmin cleavage between Lys135 and Lys136.72,73 Although both forms are capable of activating Plg, only the high-molecular-weight form binds to the u-PA receptor (see “Urokinase Plasminogen Activator Receptors” below). u-PA has much lower affinity for fibrin than t-PA and is an effective Plg activator both in the presence and in the absence of fibrin.74,75
Under certain conditions, proteases traditionally classified within the intrinsic arm of the coagulation cascade have been shown to be capable of activating Plg directly. These include kallikrein, factor XIa, and factor XIIa.76–78 These proteases, however, normally account for no more than 15 percent of total plasmin-generating activity in plasma.79 In addition, the membrane type 1 matrix metalloproteinase (MT1-MMP) appears to exert fibrinolytic activity in the absence of Plg and may explain the unexpectedly mild phenotype observed in Plg-deficient mice.80
Because there are no clinical examples of complete deficiency of t-PA or u-PA in humans, except for patients with deficient release in the setting of chronic renal disease and hypertension,81–83 the most compelling data regarding the physiologic functions of t-PA and u-PA come from gene disruption analysis in mice.84 Both u-PA– and t-PA–null deletion mice exhibit normal fertility and embryonic development. However, u-PA–/– mice develop rectal prolapse, nonhealing ulcerations of the face and eyelids, and occasional fibrin deposition in tissues. Although they show normal lysis rates of pulmonary clots injected via the jugular vein, endotoxin-induced microvascular thrombus formation is significantly enhanced. t-PA–deficient mice also display a normal spontaneous phenotype but have a decreased rate of lysis of artificially induced pulmonary thrombi, as well as enhanced thrombus formation, in response to injection of endotoxin. Like Plg–/– mice, mice doubly deficient in t-PA and u-PA (t-PA–/–; u-PA–/–) exhibit rectal prolapse, nonhealing ulceration, runting, and cachexia, with extensive fibrin deposition in liver, intestine, gonads, and lung. Not surprisingly, clot lysis is also markedly impaired.
The action of plasmin is negatively modulated by a family of serine protease inhibitors, called serpins (see Table 25–1).85 Serpins form an irreversible complex with the active site serine of their target protease following proteolytic cleavage of the inhibitor by the target protease. Within such a complex, both protease and inhibitor lose their activity.
A single-chain glycoprotein of Mr approximately 70,000, α2-PI is synthesized primarily in the liver, circulates in plasma at relatively high concentrations (~0.9 μM), and enjoys a plasma half-life of 2.4 days (see Table 25–1).86 This serpin contains approximately 13 percent carbohydrate by mass and consists of 452 amino acids with two disulfide bridges.87 In humans, the gene is located on chromosome 18 and contains 10 exons distributed over 16 kb of DNA.88 The promoter region of the α2-PI gene contains a hepatitis B–like enhancer element that directs tissue-specific expression in the liver.87 α2-PI is also a constituent of platelet α granules.89 Plasmin released into flowing blood or in the vicinity of a platelet-rich thrombus is immediately neutralized upon forming an irreversible 1:1 stoichiometric, lysine-binding site–dependent complex with α2-PI. Interaction with plasmin is accompanied by cleavage of the Arg364–Met365 peptide bond, and the resulting covalent complexes are cleared in the liver. Mice globally deficient in α2-PI display reduced fibrin deposition following treatment with endotoxin and enhanced lysis of injected plasma clots, but no spontaneous bleeding (see Table 25–2).90
Several additional proteins can act as plasmin inhibitors (see Table 25–1). α2-Macroglobulin is an Mr 725,000 dimeric protein synthesized by endothelial cells and macrophages and found in platelet α granules. This nonserpin inhibits plasmin with approximately 10 percent of the efficiency exhibited by α2-PI by forming noncovalent complexes with several distinct serine proteases.91 C1-esterase inhibitor can inhibit t-PA in plasma,92 and protease nexin may function as a noncirculating cell surface inhibitor of trypsin, thrombin, factor Xa, urokinase, or plasmin, resulting in protease–inhibitor complexes that are endocytosed via a specific nexin receptor.93,94 The purpose of these multiple plasmin inhibitors is to guard against premature plasmin activation and subsequent degradation of fibrinogen, until intravascular fibrin begins to appear.
Plasminogen Activator Inhibitor-1 Of the two major Plg activator inhibitors, PAI-1 is the most ubiquitous (see Table 25–1).95 This Mr approximately 52,000 single-chain, cysteine-less glycoprotein is released by endothelial cells, monocytes, macrophages, hepatocytes, adipocytes, and platelets.96–98 Release of PAI-1 is stimulated by many cytokines, growth factors, and lipoproteins common to the global inflammatory response.69,70,99,100,101 The PAI-1 gene consists of nine exons, spanning 12.2 kb on chromosome 7q21.3–q22.102 The serpin-reactive site is located at Arg346–Met347, and activity of this labile serpin is stabilized upon complex formation with vitronectin, a component of plasma and pericellular matrix.103–105
Regulation of PAI-1 gene expression is complex.106,107 The upstream regulatory region of the human PAI-1 gene contains a strong endothelial cell/fibroblast-specific element,108,109 a glucocorticoid-responsive enhancer,109 and TGF-β responsive elements.110 TGF-β is known to stimulate fos and jun, the two components of the AP1 complex, and an AP1 binding site (GGAGTCA) is located upstream of the PAI-1 cap site.111 Agents shown to enhance expression of PAI-1 at the message level, the protein level, or both, without affecting t-PA synthesis, include the inflammatory cytokines lipopolysaccharide, IL-1, tumor necrosis factor-α,69,70,99,112,113 TGF-β and basic fibroblast growth factor,71,99,110,114 very-low-density lipoprotein and lipoprotein(a),115,116 angiotensin II,117 thrombin,118,119 and phorbol esters.120 In addition, endothelial cell PAI-1 is downregulated by forskolin56 and by endothelial cell growth factor in the presence of heparin.121
PAI-1 is the most important and rapidly acting physiologic inhibitor of both t-PA and u-PA. Transgenic mice that overexpress PAI-1 exhibit thrombotic occlusion of tail veins and swelling of hind limbs within 2 weeks of birth.122 Mice deficient in PAI-1, on the other hand, exhibit normal fertility, viability, tissue histology, and development, and are resistant to endotoxin-induced thrombosis, but show no evidence of overt hemorrhage (see Table 25–2).123,124 These observations contrast with the moderately severe bleeding disorder observed in a human patient with complete PAI-1 deficiency.125
Plasminogen Activator Inhibitor-2 Originally purified from human placenta, PAI-2 is a 393-amino-acid member of the serpin family whose reactive site is the Arg358–Thr359 peptide bond126 (see Table 25–1). The gene encoding PAI-2 is located on chromosome 18q21–23, spans 16.5 kb, and contains eight exons.127 PAI-2 exists as both an Mr 47,000 nonglycosylated intracellular form and an Mr 60,000 glycosylated form secreted by leukocytes and fibrosarcoma cells. Functionally, PAI-2 inhibits both two-chain t-PA and two-chain u-PA with comparable efficiency (second order rate constants 105 M–1s–1). However, it is less effective toward single-chain t-PA (second order rate constant 103 M–1s–1) and does not inhibit prourokinase.
Significant levels of PAI-2 are found in human plasma primarily during pregnancy. The gene’s 5′-untranslated region contains a potent silencer, the PAUSE-1 element, which may be responsible for its low level of expression in nonpregnant individuals.127,128 The 3′-downstream sequences include the TTATTTAT motif, which has been identified with inflammatory mediators.129,130 In macrophages in vitro, secretion of PAI-2 is enhanced by endotoxin and phorbol esters,130,131 and dexamethasone decreases PAI-2 expression in HT-1080 cells.55
Thrombin-activatable fibrinolysis inhibitor (TAFI) is a plasma carboxypeptidase with specificity for carboxy terminal arginine and lysine residues.132 The action of TAFI eliminates binding sites for Plg and t-PA on fibrin.133 This single-chain Mr 60,000 polypeptide circulates in plasma at concentrations of approximately 75 nM, and undergoes limited proteolysis in the presence of thrombin, which leads to its activation.134–136 The profibrinolytic effect of activated protein C in plasma is a result of its ability to inactivate coagulation factors Va and VIIIa, which reduces activation of thrombin, the primary activator of TAFI.132 The profibrinolytic effect of activated protein C in an in vitro plasma-based system was TAFI-dependent,137 and in a system of purified components, TAFI has been shown to downregulate t-PA–induced fibrinolysis half-maximally at a concentration of approximately 1 nM, which is 2 percent of its concentration in plasma.138 Inhibition of either the intrinsic pathway of coagulation or TAFI itself results in a doubling of endogenous clot lysis in an in vivo rabbit jugular vein model of thrombolysis.139,140 TAFI-deficient mice display increased lysis of plasma clots and reduced injury-induced venous thrombosis (see Table 25–2).141,142 In plasma, TAFI may regulate Plg binding to both cell surface receptors and to fibrin.143
A large number of structurally diverse fibrinolytic “activation” and “clearance” receptors have been described. Here, we focus on endothelial cell activation receptors that are likely to contribute to homeostatic control of plasmin activity (see Table 25–1).2 Clearance receptors eliminate plasmin and Plg activators from the blood or focal microenvironments.
Plasminogen Receptors Proposed Plg receptors include α-enolase, glycoprotein IIb/IIIa complex, the Heymann nephritis antigen, amphoterin, the annexin A2/S100A10 complex, histone H2B, and plasminogen receptor-KT (Plg-RKT)2,3; these are expressed on a wide spectrum of cells, including monocytoid cells, platelets, renal epithelial cells, neuroblastoma cells, endothelial cells, and tumor cells.144–151 Typically, Plg receptors interact with the kringle structures of Plg through carboxyl-terminal lysine residues that are either present on the native protein or generated by limited proteolysis.144
Urokinase Plasminogen Activator Receptor The u-PA receptor (uPAR) is expressed on monocytes, macrophages, fibroblasts, endothelial cells, and many tumor cells (see Table 25–1).152,153 uPAR complementary DNA (cDNA) was cloned and sequenced from a human fibroblast cDNA library154 and encodes a protein of 313 amino acids with a 21-residue signal peptide. The gene consists of seven exons distributed over 23 kb of genomic DNA and places this glycoprotein within the Ly-1/elapid venom toxin superfamily of cysteine-rich proteins.155,156 uPAR is anchored to the plasma membrane through glycosylphosphatidylinositol linkages.157 u-PA bound to its receptor maintains its activity and susceptibility to the physiologic inhibitor, PAI–1.158 Formation of u-PA–PAI-1 complexes hastens clearance of u-PA by hepatic or monocytoid cells.158–161
Although originally thought to function only as a means of localizing Plg activation to the cell surface, uPAR now appears to play a central role in cellular signaling and adhesion events.152,162 The uPAR-deficient mouse has normal development and fertility and unimpaired fibrin clot lysis (see Table 25–2).163,164 uPAR binds the adhesive glycoprotein vitronectin at a site distinct from the u-PA binding domain,165,166 and u-PA transfected renal epithelial cells acquire enhanced adhesion to vitronectin while they lose their adhesion to fibronectin.167 uPAR, furthermore, colocalizes with integrins in focal contacts and at the leading edge of migrating cells,168 and also associates with caveolin, a major component of caveolae, structures abundant in endothelial cells and thought to participate in signaling events.169–171 In addition, cleaved and soluble forms of uPAR have recently been detected in the sera of patients with cancer, and these modified forms are thought to regulate the activity of several receptors involved in inflammatory and angiogenic responses.153
The Annexin A2–S100A10 System Annexin A2, an Mr 36,000, 339-amino-acid member of the annexin superfamily of calcium-dependent, phospholipid-binding proteins, forms a heterotetramer with the S100 family protein, S100A10 (see Table 25–1).172–174 It is highly conserved and abundantly expressed on endothelial cells,175–178 monocyte/macrophages,179,180 early myeloid cells,181 developing neuronal cells,182 and some tumor cells.183–185 All of the more than 60 annexin family members have in common a conserved membrane-binding C-terminal “core” region and a more variable N-terminal “tail.”186 The human annexin A2 gene consists of 13 exons distributed over 40 kb of genomic DNA on chromosome 15 (15q21).187
Annexin A2 is unique among fibrinolytic receptors in that it possesses binding affinity for both Plg (Kd 114 nM)148 and t-PA (Kd 30 nM), but not u-PA.149 In a fluid phase system of purified proteins, native human annexin A2 stimulates the catalytic efficiency of t-PA–dependent Plg activation by 60-fold.188 This effect is completely inhibited in the presence of lysine analogues or upon treatment of annexin A2 with carboxypeptidase B, an agent that removes basic carboxyl-terminal amino acids. Although it lacks a classical signal peptide, annexin A2 is constitutively translocated to the endothelial cell surface within 16 hours of its biosynthesis. This translocation event can be stimulated either by thrombin or by heat stress, in a process that requires phosphorylation of annexin A2 at Tyr23, the action of a Src family kinase, and the presence of the annexin A2 binding protein p11 (S100A10).189
At the cell surface, A2 binds phospholipid via core repeat 2, which contains the linear amino acid sequence KGLGT and downstream aspartate residue (Asp 161); together these moieties constitute a classical “annexin” motif.190 The annexin A2 heterotetramer, which consists of two A2 monomers and two protein p11 subunits and constitutes the cell surface form of A2, appears to have even greater stimulatory effects on t-PA–dependent plasmin generation.177 Interestingly, A2 regulates endogenous levels of protein p11 in the endothelial cell by masking a polyubiquitination site on p11, which otherwise directs p11 to the proteasome where it is rapidly degraded.191
Plg and t-PA appear to bind to distinct domains. Lys307 appears to be crucial for the effective interaction of Plg with annexin A2 and may be revealed upon limited proteolysis of the parent protein.188 The atherogenic low-density lipoprotein (LDL)-like particle, lipoprotein(a), competes with Plg for binding to annexin A2 in vitro,192 thereby reducing cell surface plasmin generation. t-PA binding to annexin A2 requires a domain consisting of residues 8 to 13 (LCKLSL) within the receptor’s amino terminal “tail” domain.193 This region is a target for homocysteine (HC), a thiol-containing amino acid that accumulates in association with nutritional deficiencies of vitamin B6, vitamin B12, or folic acid, or in inherited abnormalities of cystathionine β-synthase, methylenetetrahydrofolate reductase, or methionine synthase,194 and is associated with atherothrombotic disease.195,196 In vitro, HC impairs t-PA–dependent plasmin generation at the endothelial cell surface by approximately 50 percent197 by forming a covalent derivative with Cys,197 and mice with diet-induced hyperhomocysteinemia have deficient annexin A2 function198 The half-maximal dose of HC for inhibition of t-PA binding to annexin A2 is approximately 11 μM HC, a value close to the upper limit of normal for HC in plasma (12 μM).
The important role of S100A10 in fibrin balance has recently been underscored. S100A10–/– mice display increased deposition of fibrin in the vasculature and reduced clearance of batroxobin-induced vascular thrombi, and S100A10-deficient endothelial cells demonstrate a 40 percent reduction in Plg binding and plasmin generation in vitro (see Table 25–2).199 S100A10 also appears to contribute to Plg-dependent macrophage invasion in vitro by enhancing plasmin-dependent activation of matrix metalloporetinase-9.200
Several studies suggest a physiologic role for the annexin A2 system in fibrin homeostasis. First, blast cells from human patients with acute promyelocytic leukemia overexpress annexin A2 in proportion to their degree of hyperfibrinolytic coagulopathy181; S100A10 also appears to be upregulated by the PML-RAR-α oncoprotein,201 and both annexin A2 and S100A10 are downregulated by treatment with all-trans-retinoic acid. Second, in rats, arterial thrombosis can be significantly attenuated by pretreatment with intravenous annexin A2.202 Third, the prevalence of high-titer anti–annexin A2 antibodies correlates with a history of severe thrombosis in humans with antiphospholipid syndrome and in a cohort of individuals with cerebral venous thrombosis.203,204 Finally, mice with total deficiency of annexin A2 display impaired clearance of artificial arterial thrombi, fibrin deposition in the microvasculature, and angiogenic defects in a variety of tissues (see Table 25–2).205
Clearance of serpin–enzyme complexes, such as t-PA–PAI-1 and u-PA–PAI-1, occurs mainly in the liver and is mediated by a large two-chain receptor called the LDL receptor–related protein 1 (LRP1).206,207 LRP1 binds a large number of serpin–protease complexes and other ligands, indicating a multifunctional role in mammalian physiology. An additional Mr 39,000 “receptor-associated protein” copurifies with LRP1 and appears to regulate the binding and uptake of LRP1 ligands.208 Interestingly, LRP1 “knockout” embryos undergo developmental arrest by 13.5 days after conception, suggesting that regulation of serine protease activity may be crucial for early embryogenesis.209,210 Although PAI-1–independent clearance pathways for t-PA have been proposed involving the mannose receptor211 or an α-fucose–specific receptor,212 in vivo studies in mice suggest that LRP1 and the mannose receptor play a dominant role in t-PA clearance.213
Plasmin releases carboxyl-terminal Aα and N-terminal fibrinopeptide B moieties from fibrinogen (Fig. 25–3). This reaction is distinct from the proteolytic cleavage of fibrinogen by thrombin, which releases fibrinopeptide A, exposing the Gly–Pro–Arg tripeptide sequence and allowing fibrinogen to polymerize and form insoluble fibrin.214 Plasmin cleavage of fibrinogen (Mr 340,000) initially produces carboxyl-terminal fragments from the α chain within the D domain of fibrinogen (Aα fragment).205–208,215–218 Simultaneously, but more slowly, the N–terminal segments of the β chains are cleaved, releasing a peptide containing fibrinopeptide B. The resulting Mr approximately 250,000 molecule is termed fragment X and represents a clottable form of fibrinogen. Additional cleavage events may release the Bβ fragment from the β chain’s carboxyl-terminus, and in a series of subsequent reactions, plasmin cleaves the three polypeptide chains that connect the D and E domains giving rise to free D domain (Mr ~100,000) plus the binodular D–E fragment known as fragment Y (Mr ~150,000). Finally, domains D and E are separated from each other, and some of the N-terminal fibrinopeptide A sites on domain E are also modified. Although fragment X can be converted to fibrin by thrombin, the fragments Y, D, and E are all nonclottable and, in fact, may inhibit polymerization of fibrinogen.219
Figure 25–3.
Degradation of fibrinogen and crosslinked fibrin by plasmin. Top panel: On fibrinogen, plasmin initially cleaves the C-terminal regions of the α and β chains within the D domain, releasing the Aα and Bβ fragments. In addition, a fragment containing fibrinopeptide B (FPB) from the N-terminal region of the β chain is released giving rise to the intermediate fragment known as “fragment X.” Subsequently, plasmin cleaves the three connecting polypeptide chains connecting D and E domains, giving rise to fragments D, E, and Y. Bottom panel: Upon polymerization by thrombin, fibrinogen forms fibrin. When degrading crosslinked fibrin, plasmin initially cleaves the C-terminal region of the α and β chains within the D domain. Subsequently, some of the connecting regions between the D and E domains are severed. Fibrin is ultimately solubilized upon hydrolysis of additional peptide bonds within the central portions of the coiled–coil connectors, giving rise to fibrin degradation products such as D-dimer. (Reproduced with permission Nathan DG, Orkin SH, Ginsburg D, et al: Hematology of Infancy and Childhood, 6th ed. WB Saunders, Philadelphia, 2003.)
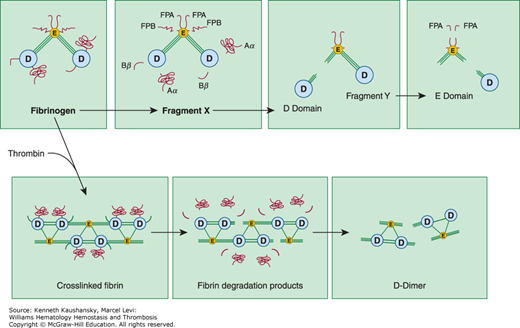
Plasmin degradation of fibrin leads to a distinct set of molecular products (see Fig. 25–3).220 Species similar to fragments Y, D, and E, but lacking fibrinopeptide sites, are released from noncrosslinked fibrin. If fibrin has been extensively crosslinked by factor XIII, however, the resulting D fragments are crosslinked to an E domain fragment. Assay of crosslinked D-dimer fragments is employed clinically to identify disseminated intravascular coagulation–like states associated with excessive plasmin-mediated fibrinolysis. Several biologic activities, including inhibition of platelet function,221 potentiation of the hypotensive effects of bradykinin,222 chemotaxis,223 and immune modulation,224 have been ascribed to fibrin breakdown products.
With or without fibrin, t-PA–mediated activation of Plg follows Michaelis–Menten kinetics.22 In the absence of fibrin, t-PA is a weak activator of Plg. However, in the presence of fibrin, the catalytic efficiency (kcat/Km) of t-PA–dependent Plg activation is enhanced by approximately 500-fold. This is the basis for its specificity as a lytic agent in the treatment of thrombosis. The affinity between t-PA and Plg in the absence of fibrin is low (Km 65 μM), but increases significantly in its presence (Km 0.16 μM), even though the catalytic rate constant remains essentially unchanged (kcat ~0.05 s–1). When plasmin forms on the fibrin surface, both its lysine binding sites and its active site are occupied. Thus, it is relatively protected from its physiologic inhibitor, α2–PI.225
The interaction of t-PA with fibrin is probably initiated by its “finger” domain. However, once fibrin is modified by plasmin, carboxy-terminal lysine residues are generated, and these become binding sites for “kringle” 2 of t-PA and “kringles” 1 and 4 of Plg.226 Therefore, fibrin accelerates its own destruction by (1) enhancing the catalytic efficiency of plasmin formation by t-PA, (2) protecting plasmin from its physiologic inhibitor, α2-PI, and (3) providing new binding sites for Plg and t-PA once its degradation has begun.
For the activation of Glu–Plg by u-PA in a fibrin-free system, reported Michaelis constants (Km) vary from 1.4 to 200 μM, while catalytic rate constants (kcat) range from 0.26 to 1.48 s–1.1 Interestingly, activation of Glu–Plg by two-chain u-PA is increased in the presence of fibrin by approximately 10-fold even though u-PA does not bind to fibrin.227 In contrast, single-chain u-PA has considerable fibrin specificity. This may reflect neutralization by fibrin of components in plasma that impair Plg74 and also reflect a conformational change in Plg upon binding to fibrin.228 It is important to recognize, however, that the intrinsic Plg activating potential of single-chain u-PA is less than 1 percent of that of two-chain u-PA. Two-chain u-PA has been used effectively as a thrombolytic agent for many years.229
A large number of in vitro studies suggest a role for plasmin in tissue remodeling. Basement membrane proteins such as thrombospondin,230 laminin,231 fibronectin,232 and fibrinogen,233 are readily degraded by plasmin in vitro, suggesting possible roles in inflammation,234 tumor cell invasion,235 embryogenesis,236 ovulation,237 neurodevelopment,238,239 and prohormone activation.240,241 Plasmin also activates MMPs 3 and 13 in the mouse, thereby facilitating the degradation of matrix proteins such as the collagens, laminin, fibronectin, vitronectin, elastin, aggrecan, and tenascin C.242 On the other hand, activation of other MMPs apparently proceeds in the absence of Plg, possibly providing the basis for the mild phenotype observed in Plg-null homozygote animals.80
Roles for plasmin in tissue remodeling and host defense mechanisms are further supported by in vivo observations in Plg-deficient mice (see Table 25–2). Impaired wound healing is observed in the Plg “knockout”243 and is reversed upon simultaneous deletion of fibrinogen.244 Plg-deficient mice also display diminished recruitment of monocytes in response to intraperitoneal thioglycolate245 and impaired neointima formation following electrical injury to blood vessels.246 In studies involving Borrelia burgdorferi, the agent of Lyme disease, dissemination of the spirochete within its arthropod vector Ixodes dammini is absolutely dependent upon host Plg even though the deer tick contains no fibrin.247 Furthermore, kainate-induced excitotoxicity and attendant neuronal cell dropout in the hippocampus is not observed in Plg knockout mice but does occur in fibrinogen-deficient animals.248 The latter two studies may define new roles for plasmin, which appear to be unrelated to degradation of fibrin.
In the lung, the fibrinolytic system mediates lung matrix remodeling through mechanisms that appear to be independent of fibrin degradation.249 In mice, deficiency of fibrinogen has no effect on the development of bleomycin-induced pulmonary fibrosis.250 Mice lacking either PAI-1 or TAFI are protected from lung fibrosis in the same model,251–253 whereas inducible expression of u-PA within alveoli abrogates the fibrotic response.254
Plasmin may play a role in the activation of growth factors. TGF-β is an Mr 25,000 homodimeric polypeptide that regulates vascular cell responses and epithelial–mesenchymal transformation in development and in tissue fibrosis.255,256 In culture, cell-associated plasmin appears to convert latent TGF-β to its physiologically relevant active state. Inhibition of wound healing in this system was dependent upon active TGF-β, and activation of this agent could be blocked in the presence of plasmin inhibitors such as aprotinin or α2-PI. Activation of TGF-β by plasmin may reflect alteration of its tertiary structure upon cleavage of an aminoterminal glycopeptide.257 Once activated by plasmin, TGF-β can stimulate production of PAI-1, thus impairing further activation of Plg.
The role of the fibrinolytic system in vascular remodeling during atherosclerosis appears to be complex.258 In the evolution of an injury to the endothelial cell lining of blood vessels, deposition of intravascular fibrin and organization of a thrombus occur.259 As the injury resolves, fibrin participates in plaque growth and luminal narrowing. Evidence of the importance of fibrinolytic balance in this process is that, in the absence of PAI-1, there is less neointima formation and reduced luminal stenosis, possibly because of more rapid resolution of fibrin.260 In areas of the vasculature where injury is not associated with fibrin deposition, however, absence of PAI-1 may lead to enhanced lesion formation, as cells that invade the developing plaque may require plasmin activity for their directed migration.261
Although the fibrinolytic system has generally been assumed to be proangiogenic by virtue of its ability to promote “tunneling” of endothelial cells through fibrin-containing matrices, its effect, in actuality, appears to be context specific.262,263 PAI-1 deficiency in mice, for example, seems to prevent tumor vascularization in a malignant keratinocyte model.264 The same mice are also resistant to laser-induced neovascularization of the choroid.265,266 The paradoxical proangiogenic effect of PAI-1 in some settings may relate to its ability to protect endothelial cells from apoptosis mediated by FasL, which is activated by plasmin.267
In the mouse cornea, absence of t-PA, u-PA, or TAFI, had no effect on neovascularization, whereas loss of Plg, PAI-1, or annexin A2 significantly diminished this response.205,268 Within the atherosclerotic plaque, moreover, expression of a truncated form of PAI-1 (rPAI-123) was antiangiogenic, inhibiting the proliferation of vasa vasorum and reducing overall plaque area and plaque cholesterol in the descending aorta.269 As a gene product that is transcriptionally upregulated by hypoxia, annexin A2 is required for the normal corneal angiogenic response to growth factor stimulation and also for hypoxia-induced retinal angiogenesis.198,205,270
Although partial human Plg deficiency was first described in a young man with a history of venous thrombosis and pulmonary embolism,271 there is currently little evidence that hypoplasminogenemia alone is a significant cause of deep venous thrombosis.272 In a study of 23 consecutive patients with thrombophilia, the prevalence of Plg deficiency was only 1.9 percent.273 Approximately half of these individuals had other risk factors such as deficiency of antithrombin, protein C, or protein S, or resistance to activated protein C. Among 93 patients with type I Plg deficiency, the prevalence of thrombosis was 24 percent, or 9 percent when the propositi were excluded.274 Two additional epidemiologic studies concluded, moreover, that isolated hypoplasminogenemia is not a risk factor for thrombosis.275,276
Although there are no reported cases of complete absence of Plg in humans, a large number of Plg polymorphisms and dysplasminogenemias have been reported.272 Congenital Plg deficiency has been classified into two types: in type I, the concentration of immunoreactive Plg is reduced in parallel with functional activity,277 whereas in type II (dysplasminogenemia), immunoreactive protein is normal while functional activity is reduced.278 Patients with type I Plg deficiency are most likely to present with ligneous conjunctivitis, which resolves completely upon infusion of Lys–Plg.279,280 In a study of a Japanese cohort, approximately 27 percent of individuals with type II deficiency had a clinical history of thrombosis, but it is not clear whether there were other explanations for thrombophilia in these individuals.281 Acquired Plg deficiency may occur in liver disease, sepsis, and Argentine hemorrhagic fever due to decreased synthesis and/or increased catabolism,282 but associated thrombosis may be due to abnormalities in other hemostatic factors in these very ill patients. Similarly, there are no reported cases of complete t-PA or u-PA deficiency in humans, and no mutations or polymorphisms in these genes have so far been clinically linked to thrombophilia. Defects in Plg activator release, as well as increased inhibition of t-PA by PAI-1, have been reported in associated with thrombosis283,284 and with chronic renal disease and hypertension.81,83
Global deficiency in fibrinolytic function, moreover, is associated with increased risk for venous thrombosis, as well as first myocardial infarction in young men.285,286 Increased circulating PAI-1 appears to represent an independent risk factor for vascular reocclusion in young survivors of myocardial infarction.287 In addition, increased levels of PAI-1 have been associated with deep vein thrombosis in patients undergoing hip replacement surgery288 and in individuals with insulin resistance.289 Although a 4G versus 5G polymorphism in the PAI-1 promoter has been reported, with the 4G form being associated with higher PAI-1 plasma levels, it is not yet established as to whether this allele correlates with elevated thrombotic risk.290,291 With regard to such studies, one should bear in mind that PAI-1 is itself an acute-phase reactant and thus may not be directly responsible for the observed prothrombotic tendency.292
Enhanced fibrinolysis resulting from congenital or acquired loss of fibrinolytic inhibitor activity may be associated with a bleeding diathesis.293 Patients with congenital deficiency of α2-PI may present with a severe hemorrhagic disorder as a result of impaired inactivation of plasmin and premature lysis of the hemostatic plug.294 Acquired α2-PI deficiency may be seen in patients with severe liver disease from decreased synthesis, disseminated intravascular coagulation from consumption, nephrotic syndrome from urinary losses, or during thrombolytic therapy, which induces excessive utilization of the inhibitor.294 TAFI levels are markedly reduced in liver cirrhosis, correlating with enhanced plasma fibrinolysis and serving as an independent predictor of mortality.295
Patients with acute promyelocytic leukemia demonstrate excessive expression of annexin A2 on their developmentally arrested promyelocytes. Bleeding in this disorder is accompanied by evidence of high levels of plasmin generation and depletion of α2-PI. Bleeding resolves upon initiation of all-trans-retinoic acid therapy, which eliminates expression of promyelocyte annexin A2, probably through a transcriptional mechanism.181 In this setting, A2 acts most likely in concert with S100A10, which is also upregulated in an acute promyelocytic leukemia (APL) cell line.201,296
Complete loss of PAI-1 expression resulting in hemorrhage in a 9-year-old child was associated with severe hemorrhage in the setting of trauma or surgery.297 This autosomal recessive trait reflected a frameshift mutation within exon 4 that induced a premature stop codon. This case demonstrates that PAI-1 is a central regulator of fibrinolysis in humans.
In the resting, nonstressed state, the plasmin-generating potential in the newborn is significantly less than that of the adult.298,299 Although the amino acid composition and apparent molecular mass of neonatal Plg are indistinguishable from those of the adult protein,300,301 plasma concentrations of Plg in the neonate are approximately 50 to 75 percent of those observed in adults.300,302,303 On the other hand, levels of histidine-rich glycoprotein, a carrier protein that may limit Plg’s interaction with fibrin, are also reduced by 50 to 80 percent in healthy, term newborns.304 Neonatal Plg is heavily glycosylated, less readily activated by t-PA, and only weakly bound to the endothelial cell surface.301 Throughout childhood, global plasma fibrinolytic activity and plasmin generation are decreased in comparison to adults, and this relative deficiency may contribute to the high frequency of thrombosis associated with central venous line placement, Kawasaki disease, and Henoch-Schönlein purpura in this age group.305
Although t-PA antigen and activity levels are reduced by 50 to 75 percent compared with adult values throughout childhood,303 stressed infants, such as those with severe congenital heart disease or respiratory distress syndrome, may have t-PA antigen levels that are increased by up to eightfold because of the t-PA release response.306,307 In contrast, the principal plasmin inhibitors undergo only minimal change from birth to adulthood.302,308–310 Thus, reduced fibrinolytic activity may contribute to the thrombogenic state commonly observed in the newborn,311 but this predilection may be reversed under conditions of physiologic stress.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


