Reference
Gender
Age
FASD
Cancer type
[7]
M
28 months
Yes
Neuroblastoma
[11]
F
3 years 9 months
Yes, with hydantoin
Hodgkin disease
[33]
F
12years 11 months
Yes
Adrenal carcinoma
[37]
M
27 months
Yes
Hepatoblastoma
[38]
M
21 months
Yes
Malignant disease
[39]
25 months
Yes
Ganglioneuroblastoma
[40]
M
110 days
Yes
Neuroblastoma Paravertebral found near kidney
[63]
M
35 months
Yes, and hydantoin
Neuroblastoma
[72]
M
35 months
Yes, and hydantoin
Ganglioneuroblastoma
[83]
M
21 months
Yes
Rhabdomyosarcoma
F
6 years
Yes
Nephroblastoma
F
16 months
Yes
Leukemia
23.2 Fetal Alcohol Promotion of Prostate Cancer
We recently tested whether an analogous circumstance occurs in the prostate because diet and hormones, especially estrogen and androgen, are crucial and interactive players in many biological and pathological processes of tumorigenesis in breast and prostate. We found that prostates of noncarcinogen-treated animals which were alcohol (6.7 % v/v) exposed during the prenatal period (day 11–21) demonstrated inflammatory cell infiltration and epithelial atypia and increased number of proliferative cells in the ventral lobe of this gland, but the prostate of control animal showed normal cytoarchitecture [46]. Prenatally ethanol exposed rats, when treated with a carcinogen N-nitrosomethylurea (NMU) and testosterone, showed histological evidence for high-grade prostatic intraepithelial neoplasia (PIN) primarily in the ventral prostate, whereas control animals showed only low-grade PIN. Prenatally ethanol exposed rats treated with NMU and testosterone also showed increased number of proliferative cells in the ventral prostate (Fig. 23.1; [46]). These results suggest that prenatal ethanol exposure induces histophysiological changes in the prostate as well as it increases the susceptibility of the prostate to develop neoplasia during adulthood.
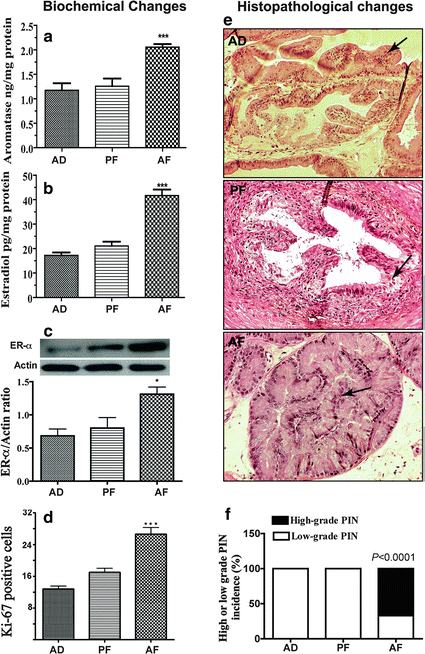
Fig. 23.1
Fetal alcohol exposures increase estrogen production in the prostate gland and promote prostate’s susceptibility to carcinogen. (a–c) Showing changes in aromatase, estradiol, and estrogen receptor (ER)-α in the ventral prostatic lobe from prenatally fed with control diet (AD, PF) or alcohol diet (AF) and sacrificed at adult age. The mean + SEM tissue concentration of aromatase and estradiol in the ventral prostate measured using ELISA are shown as histograms (a, b, respectively). Immunoblot of ER-α and actin were presented in represented gel blots and mean + SEM values as histograms (c). N = 6–7 rats. ***, P < 0.001 vs. AD and AF. (d–f) Changes in Ki-67 immunostaining and histopathology in ventral prostatic lobes of adult (7–8 months of age) fetal alcohol exposed offspring treated with NMU and testosterone (treatment details are described in [46]). The average number of Ki-67 from 5 representative slides containing 3 serial sections from each part of the ventral lobe in each group was presented in histograms (d). Representative H&E-stained photomicrographs of ventral prostatic lobes of AD, PF, and AF rat offspring (e). The ventral lobe of AD and PF rats showed low-grade prostatic intraepithelial neoplasia (PIN; arrows), while similar prostatic lobe of AF rats showed high-grade PIN. All the images were captured at 40× magnification. Mean + SEM values of percentage ratio of low-grade and high-grade PIN in animals after carcinogen treatment in AD, PF, and AF groups are shown as histograms (f). N = 9–14 rats. ***, P < 0.001 vs. AD and AF. Adopted from Murugan S, Zhang C, Mojtahedzadeh S, Sarkar DK (2013) Alcohol Clin Exp Res doi: 10.1111/acer.12171
23.3 Fetal Alcohol, Prostate Estrogenization, and Cancer
23.3.1 Evidence for an Increased Estrogen Production in the Prostate
Prostate gland development from urogenital sinus (UGS) during fetal life occurs at around 10–12 weeks of gestation in humans and 18.5 embryonic days in rats [57]. The proper development of prostate is largely dependent on the constant supply and binding of circulating testosterone or its more potent metabolite, 5α-dihydrotestosterone (DHT), to androgen receptors in the UGS mesenchyme and further activation of UGS epithelium by mesenchyme. A variation in androgen synthesis and secretion and/or delivery to the UGS has the potential to perturbate normal development [57]. Studies have shown that estrogens are equally involved in the normal and abnormal growth of prostate in human and many rodents [10, 49]. It has been shown that exposure to estrogenic compounds during the neonatal period leads to permanent alterations in prostatic growth and also produced lobe-specific histological changes later in life [22].
Neonatal exposure of rodents to high doses of estrogen permanently imprints the growth and function of the prostate and predisposes the gland to hyperplasia and severe dysplasia analogous to PIN during aging [22]. Because prostatic levels of steroid hormones and their receptors are known to be critically involved in prostate maintenance and pathogenesis [65], we measured the levels of aromatase, an enzyme essential for production of estradiol from testosterone, estrogen, and estrogen receptor-α (ER-α) in the ventral lobe of the prostate of alcohol-fed and control fed rats. We found that the aromatase activity, ER-α immunoreactivity, and estrogen level were higher in the ventral prostate of fetal alcohol exposed offspring (Fig. 23.1). These findings indicate that the prostate gland of fetal alcohol exposed rats produce more estrogen than the control rats.
The initiation of prostatic development is known to be dependent on androgens produced by the fetal testes [54]. In addition, the developing prostate is particularly sensitive to estrogens [55, 67]. Estrogen receptor α (ERα) localizes to proximal mesenchymal cells during early development in rodents and declines as morphogenesis proceeds implicating a specific developmental role for estrogens [55]. ERβ is induced in luminal epithelial cells upon cytodifferentiation suggesting a role for ERβ in differentiated function [41, 58]. In humans, under the influence of maternal estrogens, all fetal males contain marked prostatic squamous metaplasia which sloughs at birth [87]. Additionally, maternal exposure to pharmacological levels of diethylstilbestrol (DES) has been shown to induce prostatic abnormalities in human offspring [23].
Consequently, it is proposed that excessive estrogenization during prostatic development may contribute to the high incidence of prostatic carcinoma observed in the aging male population [67]. Previous works from various laboratories addressing this hypothesis have documented that brief exposure of rodents to high doses of natural or synthetic estrogen early in life results in permanent alterations of the prostate gland, a phenomenon referred to as estrogen imprinting or developmental estrogenization [55, 59, 62]. Organ culture studies confirm a direct effect since the developmental aberrations are recapitulated upon estrogen addition [61]. Using the Sprague–Dawley rat model, it has been shown that brief exposure to high levels of estrogens during the neonatal period (days 1–5) causes marked developmental and cellular differentiation defects [56], dose-dependent reductions in adult prostate size [29, 48, 62], and compromised secretory capacity in adulthood [31, 62]. Upon aging, prostatic inflammation, epithelial hyperplasia, PIN, and eventually adenocarcinoma are prominent [55]. Thus, it appears that developmental estrogen exposures may predispose to neoplasia in adulthood.
23.3.2 Connection between the Developmental Estrogenization and Prostatic Neoplasia
Alcohol consumption has been shown to be associated with a 20 % increase in serum estrogen levels in women [43], and this finding has been supported by rodent studies [32]. Exposure to estrogens in utero [14], or synthetic estrogen diethylstilbestrol (DES) treatment has also been associated with a higher risk for breast cancer in female offspring and prostate cancer in male offspring later in life [23]. It is interesting to note that although the initiation of prostatic development is dependent on androgens produced by the fetal testes [54], the developing prostate is particularly sensitive to estrogens [55, 67]. In the estrogen receptor (ER) positive cell line MCF-7, alcohol treatment resulted in a dose-dependent increase in cell proliferation. In contrast, no increase in proliferation was observed in ER negative cell lines. In the same study, it was shown that alcohol also induced production of ERα protein in a dose-dependent manner [74]. These data suggest that ER-α may be one of the factors involved in alcohol-induced tumorigenesis. It is also interesting to note that combined treatment of estrogen and testosterone induces biochemical and histological carcinogenesis in ER-β-knockout (β-ERKO) mice but not in ER-α-knockout (α-ERKO) mice, suggesting a role for ER-α in prostate carcinogenesis [65]. In humans, under the influence of maternal estrogens, all fetal males contain marked prostatic squamous metaplasia which sloughs at birth [87]. Additionally, maternal exposure to pharmacological levels of diethylstilbestrol (DES) has been shown to induce prostatic abnormalities in human offspring [23].
Consequently, it is proposed that excessive estrogenization during prostatic development may contribute to the high incidence of prostatic carcinoma observed in the aging male population [67]. In this regard, fetal exposure to alcohol has been shown to alter pubertal mammary gland development [53] due to excessive estrogenization [32, 52] and increases aromatase and ER-a levels in prostate [46]. Furthermore, prolonged adult exposure to estradiol at levels within a physiologic range is capable of driving prostatic carcinogenesis in the Noble rat model [82]. Thus, the possibility exists that developmental estrogenic exposures within the prostate may lead to cellular abnormalities and carcinogenesis in the adult life.
23.4 Fetal Alcohol, Neuroimmune Axis Abnormalities, and Cancer
23.4.1 Neuroendocrine–Immune System and Tumor Surveillance
As discussed earlier fetal alcohol exposure increases the incidence of various hormone-dependent and independent cancers. The question arises why the fetal alcohol exposed offspring shows higher tumor incidence in many tissues? It could be hypothesized that many types of cancers in fetal alcohol exposed patients might have been promoted due to an abnormality in the physiological process(s) that prevents cancer development as well as molecular processes (e.g., prostate estrogenization) that control growth and differentiation of a specific cell population. There are two biological systems that are significantly altered in fetal alcohol exposed patients. One relates to stress regulation and the other is the immune system function. Children who are exposed to alcohol during fetal life often show behavioral and physiological changes such as depression, anxiety, hyperactivity, and an inability to deal with stressful situations [30, 75]. Fetal alcohol exposed patients and prenatal ethanol exposed rats often show elevated basal and stimulated levels of the adrenal hormone glucocorticoid, pituitary hormone adrenocorticotropic hormone (ACTH), and hypothalamic hormone corticotropin-releasing hormone (CRH) in response to stressors such as repeated restraint, foot shock, and immune challenges [13, 42, 85]. Children prenatally exposed to alcohol have lower cell counts of eosinophils and neutrophils, decreased circulating E-rosette-forming lymphocytes, reduced mitogen-stimulated proliferative responses by peripheral blood leukocytes, and hypo-c-globulinemia [35]. In animal models, fetal alcohol exposure is shown to negatively affect lymphoid tissue development, immune cell function, humoral immunity, and cytokine productions [4]. Activity of NK cells is also suppressed in these animals [13]. Children prenatally exposed to alcohol often have an increased incidence of bacterial infections such as urinary tract and upper respiratory tract infections [35].
Studies from human and animal models have shown that acute and chronic stressful events have adverse effects on a variety of immunological mechanisms, such as trafficking of neutrophils, macrophages, antigen-presenting cells, natural killer (NK) cells, and T and B lymphocytes [9, 16]. Exposure to stress modulates cell-mediated immunity by suppressing lymphocyte proliferation and NK activation, lowering the number of CD4+ cells in the peripheral blood and altering CD4/CD8 T cell ratios [9, 81]. Studies have also shown that depression and stress might have effects on carcinogenesis indirectly, through the poorer destruction or elimination of abnormal cells by reduced NK cell activity. Decreased NK cell activity is also associated with growth and progression of a variety of cancers in animals and humans, because NK cells appear to represent a first line of defense against the metastatic spread of tumor cells [50]. Stress is also associated with altered inflammatory and anti-inflammatory cytokine ratios in systemic circulation, increases expression of interleukin-1 beta (IL-1ß) and tumor necrosis factor-alpha (TNF-α), and reduced expression of IL-2 and interferon-gamma (IFN-γ) [2]. Sustained elevation of TNF-α is known to inhibit the activity of protein tyrosine phosphatase (PTPase), causing reduced production of the MHC class I antigen of the cell surface, and leading to malignant cells escaping immune surveillance [45]. Although there are many specific details yet to be delineated, it is becoming increasingly clear that stressful life events can impact cancer growth, progression, and metastasis by modulating nervous, endocrine, and immune systems of the body. Thus, reduced immune function and increased susceptibility to cancer may be a consequence of the stress axis abnormalities in fetal alcohol exposed offspring.
23.4.2 Neuroendocrine–Immune System Abnormalities and Prostatic Neoplasia
As discussed earlier, studies in animals indicated that fetal alcohol exposed offspring show elevated basal and hyper stress response to stressors and immune challenges [13, 35, 42, 85]. Can these changes of the stress axis relate to an abnormality in the feedback regulation of the axis? It is known that beta-endorphin (BEP), a hormone release from a set of neurons in the hypothalamus inhibits CRH secretion and downregulates the HPA axis functions. Fetal alcohol decreases the levels of BEP in the hypothalamus during adult life [17, 68]. Furthermore, transplantation of BEP neurons in the hypothalamus normalizes the HPA dysfunction in fetal alcohol exposed offspring [13]. Therefore, the stress hyperactivity in fetal alcohol exposed offspring may have resulted from a decreased BEP neuronal function Fig. 23.2.
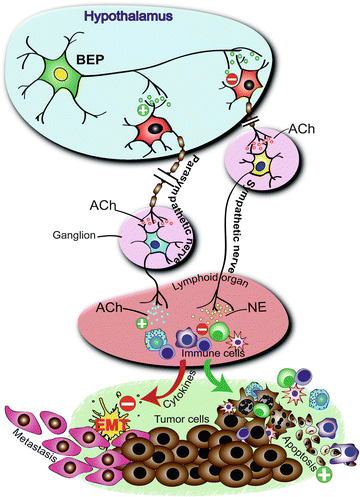
Fig. 23.2
Beta-endorphin (BEP) neuron in the hypothalamus controls the growth and progression of tumor cells by modulating the neurotransmission in the autonomic nervous system and activating innate immune system. Effects include the stimulation of parasympathetic nervous system and release of acetyl choline (Ach) and suppression of the sympathetic nervous system and release of norepinephrine (NE) leading to activation of innate immune cells (including macrophages and natural killer cells) of the lymphoid organ and an increase in cytotoxic immune cells and anti-inflammatory cytokine levels in the circulation. In a tumor microenvironment these immune cell and cytokine changes increase apoptotic death of tumor cells and reduce inflammation-mediated epithelial-mesenchymal transition (EMT), and thereby suppress cancer growth and metastasis. Collectively, these effects create an unfavorable environment for tumor initiation, growth, and progression. From: Zhang C, Sarkar DK (2012) Oncoimmunology 1(4):552-554
BEP is an endogenous opioid polypeptide, a cleavage product of proopiomelanocortin (POMC), which is also the precursor hormone for adrenocorticotrophic hormone (ACTH) and α-melanocyte-stimulating hormone (α-MSH). POMC products are produced and secreted by the hypothalamus (BEP and α-MSH) and pituitary gland (ACTH, α-MSH and BEP) in vertebrates during exercise, excitement, pain, and orgasm, and they resemble the opiates in their abilities to produce analgesia and a feeling of well-being [3, 26].
BEP neuronal cell bodies are primarily localized in the arcuate nuclei of the hypothalamus, and its terminals are distributed throughout the CNS, including the paraventricular nucleus of the hypothalamus [69]. In the paraventricular nucleus these neurons innervate CRH neurons and inhibit CRH release [36]. During stress, secretion of CRH and catecholamine stimulate secretion of hypothalamic BEP and other POMC-derived peptides, which in turn inhibit the activity of the stress system [51]. BEP is known to bind to δ- and μ-opioid receptors and modulate the neurotransmission in sympathetic neurons via neuronal circuitry within the paraventricular nucleus to alter NK cell cytolytic functions in the spleen [12, 13]. Low levels of hypothalamic BEP are correlated with a higher incidence of cancers and infections in patients with schizophrenia, depression, and fetal alcohol syndrome and in obese patients (reviewed in [84]). Hence, hypothalamic BEP inhibits CRH secretion and sympathetic outflow to the lymphoids and stimulates immunity.
We have recently shown that in vitro produced BEP neurons, when transplanted in the paraventricular nucleus of the hypothalamus, remained at the site of transplantation, and increase NK cell cytolytic function and production of anti-inflammatory cytokines in response to an immune challenge [69, 70]. BEP transplantation in the paraventricular nucleus of the hypothalamus also suppresses carcinogen-induced prostate and mammary tumorigenesis. Importantly, when the BEP transplants were given at an early stage of tumor development, many tumors were destroyed possibly due to increased innate immune activity, and the surviving tumors lost their ability to progress to high-grade cancer due to BEP cells’ suppressive effects on epithelial–mesenchymal transition (EMT) regulators. Another remarkable effect of the BEP transplantation was that it promoted the activation of the innate immune activity following tumor cell invasion to such an extent that tumor cell migration to another site was completely halted. The cellular mechanism involved in the cancer preventive effects of BEP appears to involve alteration of the autonomic nervous system functioning, leading to activation of innate immunity and reduction in systemic levels of inflammatory and anti-inflammatory cytokine ratios. Since fetal alcohol exposed animals have BEP neuronal deficiencies and higher incidence of aggressive prostate tumors following a carcinogen treatment, the possibility is raised that the stress abnormalities and resulting immune incompetence might have contributed to the increased prostatic neoplasia in these animals. Our recent preliminary data support this concept and show that BEP neural functional abnormality is also one of the causes for increased cancer incidence in fetal alcohol exposed offspring [84].
Related posts:
 A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
 Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
 Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
 Alcoholic Cirrhosis and Hepatocellular Carcinoma
Alcoholic Cirrhosis and Hepatocellular Carcinoma
 Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


