Fig. 24.1
Alcohol exposure in utero increases total number of mammary tumors in offspring. (a) Rats exposed in utero to low (7 % of total calories) or moderate (15 % of total calories) alcohol or an isocaloric liquid diet (control) were injected with DMBA at 47 days of age. Adapted by permission from Macmillan Publishers Ltd: on behalf of Cancer Research UK [24]. (b). Rats exposed to high alcohol (35 % of total calories), an isocaloric liquid diet (pair-fed), or ad libitum rat chow (ad lib-fed) were treated with NMU at approximately 50 days of age. Reproduced with kind permission from Springer Science + Business Media [131]
These studies suggest that an additional adverse outcome of alcohol consumption during pregnancy could be that women born to these mothers have increased risk of developing breast cancer as adults. Fetal exposure to alcohol has been shown to increase risk for childhood leukemia, suggesting that findings in rodent models may be relevant to human disease [27]. There is considerable discussion surrounding the over-diagnosis and overtreatment of breast atypia due to the difficulty in discerning which of these will progress to invasive cancer [28]. Therefore, it is of utmost importance to identify additional risk factors for breast cancer so that we can clearly distinguish which groups of women are at high risk for developing the disease.
While the finding that rodent offspring born to alcohol-exposed dams exhibit increased susceptibility to carcinogens as adults is consistently observed, the mechanisms underlying this increased susceptibility are not well understood. The estrogen and insulin-like growth factor (IGF) axes occupy central roles in both mammary gland development and breast cancer. This chapter will discuss evidence that this endocrine axis may be affected by alcohol exposure in utero leading to increased mammary tumorigenesis and propose potential mechanisms for these alterations.
24.2 Overview of Mammary Gland Development
The development of the mammary gland depends on complex molecular interactions between the parenchyma and the supporting stroma throughout three main stages of life: fetal development, puberty, and reproduction (pregnancy, lactation, and involution). The parenchyma consists of luminal and basal epithelial cells structured into ducts for milk production with adjacent myoepithelial cells that secrete basement membrane, while the stroma is comprised of multiple cell types including adipocytes, fibroblasts, immune cells, and nerve cells [29]. Much of what is known about human mammary gland development comes from rodent models [30].
In rats, mammary development begins between day 10 and 11 of gestation at which time the mammary streak (line) is detected and consists of a single layer of ectodermal tissue stretching from the anterior to the posterior limb buds [31]. Over the next day, epidermal cells migrate to form several layers of cells, creating the mammary placodes [32, 33]. In rats there are six pairs of mammary placodes which correspond to the number of mammary glands in the adult. Each mammary placode invaginates into the underlying mesenchyme to become a bulb-shaped mammary bud. The surrounding mesenchymal cells reorient themselves into a radial pattern surrounding the parenchyma to form a pad of fatty connective tissue (the fat pad precursor). After a 3–4 day morphological quiescence there is a period of rapid cellular proliferation, which results in the mammary sprout. This elongates, forming ducts that penetrate the fat pad precursor such that at birth, the gland is comprised of a limited number of branching ducts [31, 34, 35].
After birth the mammary gland continues to grow isometrically with the rest of the body until the onset of ovarian hormones during puberty, when growth becomes allometric. The leading edge of each duct enlarges to form a club-shaped structure called a terminal end bud (TEB), which contains highly proliferative cells. Once the ducts have penetrated the fat pad and undergone initial bifurcations, the complexity of the milk duct system increases through side branching during each estrous cycle. During pregnancy, ductal side branching becomes even more extensive. Subsequently during lactation the gland becomes mature and fully functional. The epithelial component of the gland proliferates and fills the fat pad with lobular alveolar structures. The alveoli differentiate to produce milk proteins which are secreted into the ductal lumen and transported to the nipple. Once suckling ceases the gland undergoes involution, during which epithelial cells undergo apoptosis and the alveolar structures dedifferentiate [35].
24.3 Role of Estrogen in Mammary Gland Development and Breast Cancer
Estrogens are steroid hormones that are produced mainly in the ovaries in pre-menopausal women, with minor contributions from adipose tissue and the adrenal glands. 17β-estradiol (E2) is the major circulating estrogen, but minor forms include estrone (E1) and estriol (E3), with E3 produced by the placenta during pregnancy. The hypothalamic-pituitary-gonadal (HPG) axis regulates ovarian production of estrogen. The hypothalamic peptide gonadotropin-releasing hormone (GnRH) induces release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) which simulate androgen production and aromatase activity, respectively. Aromatase catalyzes the aromatization of testosterone to E2, the final rate-limiting step of E2 biosynthesis. The high levels of E2 released during ovulation feedback to inhibit the release of GnRH, LH, and FSH.
The classical genomic mechanism of E2 action is initiated when E2 diffuses across the plasma membrane and binds to ER in the cytoplasm, where it resides bound to heat-shock proteins (Fig. 24.2). Binding of E2 to ER results in its dissociation from these proteins, leading to dimerization of the receptor and translocation to the nucleus where the complex functions as a ligand-activated transcription factor by binding to estrogen response elements (EREs) to initiate expression of multiple target genes [36–38]. Additionally, non-genomic effects of E2 have been described and will be discussed more below [39, 40].
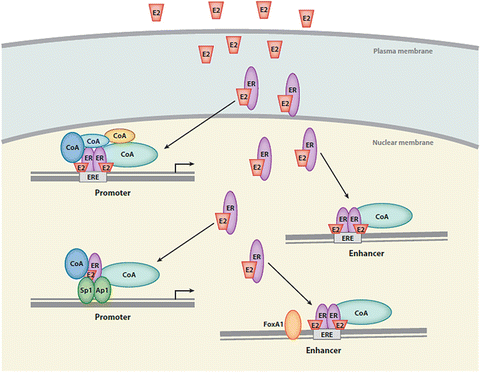
Fig. 24.2
Classical estrogen signaling pathway in the cell. 17-β estradiol (E2) diffuses across the plasma membrane and binds to ER in the cytoplasm. The E2/ER complex translocates to the nucleus and binds to estrogen response elements (EREs) to initiate gene expression. Reproduced with permission of Annual Reviews in the format Republish in a book via Copyright Clearance Center [38]
Estrogens act primarily through two nuclear ERs, ER-α and ER-β. E2 is required for the initial formation of the TEBs during pubertal mammary gland development. Ovariectomy of mice around the onset of puberty causes regression of TEBs which reappear when E2 pellets are implanted [41]. Additionally, ER-α knockout mice (ERKO) have mammary glands that appear rudimentary and lack any branching or TEBs [42]. Transplantation studies indicate that while ER-α is expressed in both the stroma and the epithelium of the developing gland [41], ER-α in the epithelial cells is most important for growth [42]. Deletion of ER-β does not affect ductal growth of the mammary gland [43] though it does interfere with terminal differentiation during lactation [44].
A role for E2 in the etiology and progression of breast cancer was first recognized when bilateral ovariectomy in pre-menopausal breast cancer patients was found to result in cancer remission [45]. This finding led to investigation into the role of ovarian hormones and their receptors in breast cancer. Since approximately 70 % of all breast cancers are ER-α positive and require E2 for growth, an effective treatment strategy in women with ER-α positive tumors has been to block the E2 system with either selective ER modulators (SERMs) that bind to and block ER-α, or aromatase inhibitors, which act by inhibiting E2 production [46]. For this reason, ER-α has become a prognostic and predictive marker for breast cancer.
The fact that a number of breast cancer risk factors correspond with an increase in exposure of the mammary gland to E2 (e.g., women who experience menarche at an early age and/or menopause at a later age) has led to the idea that lifetime exposure to this hormone is a primary risk factor for the disease [47]. Increased exposure to E2 across a women’s lifetime is thought to indirectly affect carcinogenesis by increasing the number of cell cycles and thus the chances of genetic mutations. In addition, many studies have reported that increased serum E2 is positively associated with breast cancer risk in postmenopausal women [48–50]. However, recent meta-analyses of multiple epidemiological studies suggest that postmenopausal hormone concentrations are not strongly related to age at menarche or first full-term pregnancy, parity or family history [51, 52], suggesting that these risk factors are not likely mediated through postmenopausal hormone levels. These risk factors may instead operate through long-term effects of sex hormone levels in pre-menopausal women such as changes in the duration rather than the level of long-term exposure to sex hormones, by permanent changes in breast structure induced by pregnancy and lactation, or by other mechanisms [51].
A second mechanism by which E2 may impact carcinogenesis is via direct carcinogenic properties of E2 metabolites. The conversion of E2 to 2- or 4-OH-E2 results in reactive oxygen species byproducts that are carcinogenic [53]. Also, the metabolites themselves can directly bind DNA and form adducts leading to mutagenesis [54, 55]. The ERKO/Wnt-1 transgenic mouse demonstrates the importance of these metabolites in tumor initiation. In this study treating ERKO/Wnt-1 transgenic mice with E2 increased mammary carcinogenesis in the absence of ER-α, thus implicating the metabolites as the culprits [56].
24.4 Role of the IGF System in Normal Mammary Gland Biology and Breast Cancer
The IGF family is comprised of two ligands (IGF-I and IGF-II) and six IGF-binding proteins (IGFBP-1 through IGFBP-6) that modulate the actions of IGFs. They exert their biological effects primarily through the IGF type I receptor (IGF-IR), a heterodimeric intrinsic tyrosine kinase receptor [57, 58]. The IGF–IR gene encodes a protein consisting of an extracellular α ligand binding domain and a β subunit containing the intracellular tyrosine kinase domain which dimerizes with a second unit to form the functional receptor. The insulin receptors A and B are closely related to IGF-IR and it has recently been recognized that IGF-insulin hybrid receptors may also influence IGF action in the mammary gland [59, 60]. Binding of IGF ligand induces autophosphorylation of the receptor, followed by activation of adapter molecules such as insulin-receptor substrate 1 (IRS-1) and Shc, setting in motion activation of the phosphatidyl inositol-3-kinase (PI3K) and mitogen activated protein kinase (MAPK) signal transduction cascades (Fig. 24.3). These signaling molecules activate downstream factors that stimulate multiple processes that regulate normal growth and development of the mammary gland, including cellular proliferation, survival, angiogenesis, and migration. These processes also underlie abnormal growth processes that lead to breast cancer, including tumorigenesis and metastasis.
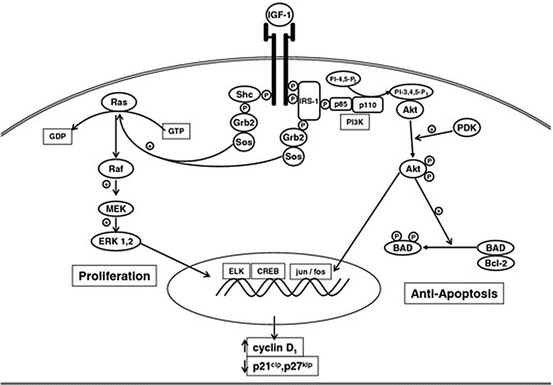
Fig. 24.3
Signal transduction pathways activated by IGF-I via the IGF-I receptor in the cell Reproduced with permission [131]
Development of the mammary gland is regulated by complex hormonal interactions involving the pituitary hormones, growth hormone (GH) and prolactin, and the ovarian hormones, estrogen and progesterone. The postnatal effects of GH in mammary gland development are predominately mediated through IGF-I [61]. The importance of IGF-I in mammary gland development is demonstrated by the finding that IGF-I knockout mice have dramatically impaired TEB formation and ductal development [62]. Understanding the role of the IGF system in normal mammary gland development is complicated by the fact that IGF-I exerts it effects on the mammary gland through both endocrine and autocrine/paracrine mechanisms [58]. The majority of circulating IGF-I is produced in the liver, while most tissues, including the mammary gland, also produce IGF-I at the local level. GH can increase circulating IGF-I by binding to hepatic GH receptors and regulating hepatic production and/or it can bind to GH receptors in the mammary gland to increase local production [63]. Mice that exhibit reduced local and circulating levels of IGF-I (IGF-I midi mice) have normal ductal elongation into the fat pad but fewer branching structures than normal mice. However, mice with a liver-specific deletion of IGF-I (LID mice) exhibit a 75 % reduction in circulating IGF-I, yet maintain normal mammary ductal branching [64], suggesting that locally produced IGF-I acts in a paracrine fashion to regulate branching morphogenesis. Studies with transgenic mice that overexpress hepatic IGF-I (HIT) but have no local IGF-I production (IGF-I knockout/HIT) show that circulating IGF-I can maintain normal mammary gland development in the absence of local IGF-I. In addition, HIT mice exhibit enhanced mammary gland proliferation compared to control mice, indicating that elevated circulating IGF-I in the presence of local IGF-I production can accelerate mammary gland growth [65]. Collectively these studies show that both local and endocrine actions of IGF-I are important in mammary gland development.
Both circulating and local production of IGF-I are also important in mammary tumorigenesis and progression. In LID mice, administration of the carcinogen DMBA results in a lower incidence of mammary tumors and delays tumor onset [66], supporting a role for circulating IGF-I in breast tumor progression. Since IGF-I is predominately made in the stromal tissue in response to GH, a transgenic model was generated that mimics paracrine exposure of breast epithelial to stromal IGF-I by placing IGF-I under control of the keratinocyte 5 promoter, leading to overexpression of IGF-I in myoepithelial cells. These mice show increased ductal proliferation prepubertally compared with wild-type mice and exhibit increased susceptibility to carcinogen-induced tumorigenesis as adults [67].
As mentioned above, the biological activity of IGF-I is modulated by a family of six high affinity IGFBPs. Of these IGFBPs, a large body of evidence supports a role for IGFBP-5 as a growth-inhibitory and/or pro-apoptotic factor in the mammary gland [68]. There is a positive relationship between IGFBP-5 expression and cell death during mammary involution. Transgenic overexpression of IGFBP-5 in mammary tissue causes increases in apoptotic death of epithelial cells and reduces invasion of the mammary fat pad, while addition of exogenous IGFBP-5 to murine mammary epithelial cells suppresses IGF-I mediated survival [69, 70]. Implantation of IGFBP-5 into one mammary gland of hypophysectomized, oophorectomized Sprague Dawley rats decreases the ability of GH and E2 to stimulate TEB formation after 7 days [71]. Likewise, when IGFBP-5 is injected into wild-type intact mice for 7 days beginning on day 16 of pregnancy, the proportion of secretory tissue in the gland is decreased and lactation is impaired [72]. Many of these pro-apoptotic, growth-inhibitory effects are thought to be related to the ability of IGFBP-5 to sequester IGF-I and prevent its pro-survival and growth-stimulatory effects.
24.5 Cross-Talk Between the E2 and IGF Systems
The E2 system interacts extensively with the IGF system in modulating mammary gland development and tumorigenesis [73–75]. During mammary gland development, administration of E2 or IGF-I alone to ovariectomized, hypophysectomized rats does not restore full ductal development; however, the gland develops normally when these hormones are administered together [76]. Likewise, both E2 and IGF-I are required to restore normal mammary gland development in the IGF-I knockout mouse [62]. At the cellular level, normal breast epithelial cells that are positive for ER-α do not stain positive for Ki67 or bromodeoxyuridine (BrdU) incorporation, indicating that E2 is not directly driving proliferation [77–79]. E2 is believed to stimulate the release of IGF-I, which acts in a paracrine manner to drive growth of adjacent cells [80].
In breast cancer cells, the proliferative effect of E2 is accounted for, in part, by its ability to upregulate components of the IGF signaling pathway, including IGF ligands, IGF-IR, and IRS-1 [81–84]. Reciprocally, IGF ligands can act via the IGF-IR to enhance transcriptional activation of ER-α by promoting its binding to EREs or by phosphorylating ER-α via activation of the PI3K and MAPK pathways [85–88]. The interactions between ER and IGF-IR may be mediated through a small pool of ERs localized to the plasma membrane, which can also occur in the absence of sex steroid [37, 89, 90]. These include not only ER-α and ER-β, but also ER-α transcript variants (including ER-α36) and the G-protein-coupled ER 1 (GPER, formerly known as GPR30), a G-protein coupled receptor that is structurally different from the classical ERs [91, 92]. A complex relationship between the membrane and nuclear effects of E2 also involves membrane-initiated phosphorylation of coactivators, which are then recruited to the nucleus. In addition to localizing to the nucleus and the plasma membrane, some ER may also localize to mitochondria [37]. Therefore, the integration of effects that are mediated by ERs at distinct cellular locations plays a major role in regulating cellular outcomes in the cell. The finding that the E2/ER/IGF systems interact so extensively has led to the development of an array of therapeutic agents to target the IGF-IR signaling system in cancer, including breast cancer. However, despite promising results in early phase trials, randomized phase III trials have not yet been successful, demonstrating the complexity of the IGF signaling system [93, 94].
24.6 Evidence for Alterations in E2 and IGF Systems in Alcohol-Exposed Offspring
Several lines of evidence from rodent studies conducted in our laboratory show that alcohol-exposed offspring exhibit alterations in various components of the E2/IGF axis. Few mutations in components of the E2/IGF axis have been reported in humans which demonstrate the primary importance of these system components to normal growth and development. Therefore it is possible that alterations in the E2/IGF system are caused by epigenetic modifications, defined as heritable but reversible changes in gene function that occur without changes in nucleotide sequence. In the sections below, we outline evidence indicating that exposure to alcohol in utero induces epigenetic events. It is plausible that there could be epigenetic changes to E2/IGF hormone action in response to these alcohol-induced modifications. This regulation could occur at one or more levels to affect hormone synthesis, circulating and/or target tissue hormone levels, and/or target-organ responsiveness [95].
To determine if prenatal alcohol exposure induces early changes in mammary gland morphology that might enhance the susceptibility to a carcinogen in adulthood, we used the same in utero model of alcohol exposure described in the introduction. Instead of injecting NMU at postnatal day (PND) 50, we euthanized animals at three developmental time points: PND 20, a prepubertal time point when the ductal structures in the mammary gland are highly proliferative, PND 40, near puberty when the HPG hormonal axis has been activated, and PND 80, when the mammary gland is mature. Offspring were injected with BrdU prior to euthanization to quantitate cell proliferation. At 20 and 40 days of age, animals exposed to alcohol in utero had an increased proliferative index compared to PF controls [26]. The increases between days 20 and 40 indicate that in utero alcohol exposure causes early changes in programming of the mammary gland that may contribute to the enhanced tumorigenesis observed in response to NMU.
Given the increase in the proliferative state of the mammary gland at this early age, we hypothesized that alterations in the IGF-I axis might contribute to this effect. Hepatic IGF-I mRNA levels were increased at all three time points in alcohol-exposed offspring, while IGF-I mRNA levels were also increased in the mammary gland at PND 20. Interestingly, IGFBP-5 mRNA levels were significantly lower at PND 40 in animals exposed to prenatal alcohol relative to pair-fed controls (Fig. 24.4). Therefore, a decrease in IGFBP-5 expression in tumors of rats exposed to alcohol in utero may allow more free IGF-I to access the IGF-IR and promote tumorigenesis. IGFBP-5 is reduced in epithelial tumors such as head and neck squamous cell carcinoma and cervical carcinoma and has been proposed as a tumor suppressor gene [96, 97]. Therefore, the increased proliferation we observed in alcohol-exposed offspring could also be related to a decrease in a tumor suppressor role of IGFBP-5, which could be independent of IGF-I.
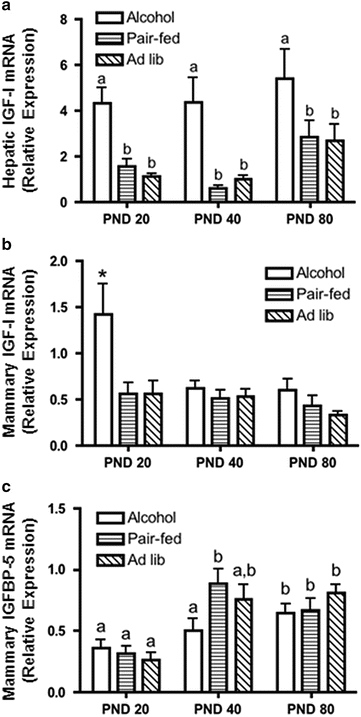
Fig. 24.4
Alcohol exposure in utero increases hepatic and mammary IGF-I expression and delays the developmental increase in mammary IGFBP-5 expression. Following alcohol exposure in utero, liver and mammary glands were collected at sacrifice on postnatal day (PND) 20, 40, or 80. RNA was isolated and analyzed by qRT-PCR. (a, c) Different letters denote significant differences (P < 0.05); (b) Asterisk denotes significance at P < 0.001. Adapted from [26] with kind permission from Springer Science + Business Media
In addition to changes in the IGF axis, aromatase expression was increased in the mammary glands of alcohol-exposed offspring at both PND 20 and 40 [26]. As mentioned above, aromatase is the key enzyme that converts testosterone to E2. Interestingly, IGF-I enhances aromatase activity in a variety of cells although the mechanism is not well understood. Recently, IGF-I was shown to increase aromatase activity by decreasing degradation of aromatase with no corresponding change in mRNA levels [98]. Therefore both IGF-I and E2 expression may be increased locally in mammary glands of alcohol-exposed offspring, where they may cross-talk to promote mammary proliferation in early development.
To further test our hypothesis that the E2 system is altered by alcohol exposure in utero we conducted another study in alcohol-exposed adult offspring in which we monitored the estrus cycle by vaginal cytology and sacrificed all animals during proestrus. Animals exposed to alcohol in utero exhibited significantly higher circulating levels of E2 relative to either pair-fed or ad libitum control groups (Fig. 24.5). These data may be related to findings that in utero alcohol exposure alters the development and maturation of the HPG axis in female rats [99].
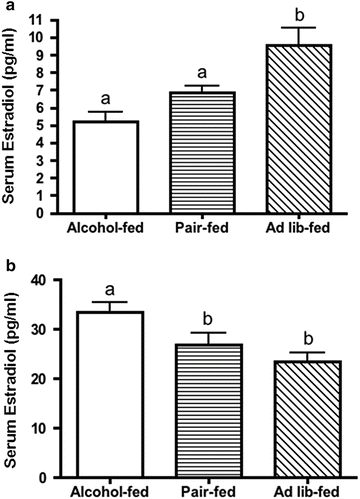
Fig. 24.5
Alcohol exposure in utero increases circulating levels of 17-β estradiol (E2) during proestrus in adulthood. Pups were sacrificed during proestrus (ages 62–76 days) to avoid cyclic differences in circulating E2 levels and trunk blood was collected. E2 levels were determined by ELISA. Different letters denote significant differences (P < 0.05). Republished from [25] with kind permission from Springer Science + Business Media
In addition to alcohol, exposure to a wide variety of toxic compounds during fetal or neonatal development affects postnatal mammary gland development and/or leads to increased susceptibility to mammary carcinogens in adulthood, supporting the fetal origins of disease hypothesis [100]. Many of these toxins are endocrine disruptors, which act by interfering with E2 action. Furthermore, rodent studies have shown that maternal exposures to dietary factors such as high fat leads to alterations in mammary morphology during development and/or an increased risk of mammary cancer in the offspring [101, 102]. Feeding a high fat diet also increases circulating concentrations of E2 in the dams [101]. Exposure to estrogens in utero due to female twin-ship [103] or DES treatment [104] is associated with a higher risk for breast cancer later in life. Based on the rationale that alterations in the fetal estrogenic environment may change the susceptibility of the mammary gland to later exposure to carcinogens, Hilakivi-Clarke et al. [24] measured E2 in the circulation of pregnant dams exposed to alcohol. They found an increase in E2 concentrations in the dams fed the lower level of alcohol (7 % of total calories) but not in the dams fed the intermediate level of alcohol (15 % of total calories). Interestingly, tumor number was only increased in the dams fed the intermediate level of alcohol. Whether circulating E2 levels are increased in dams fed high concentrations of alcohol has not yet been determined.
24.7 Fetal Alcohol and Epigenetics
As mentioned above, epigenetics involves heritable changes in gene expression that occur without an alteration in the primary nucleotide sequence of a gene. Epigenetic modifications to nuclear chromatin structure alter DNA, histones, and non-histone proteins. These modifications limit or enhance the accessibility and binding of the transcriptional machinery or recruit repressor complexes, resulting in changes in gene expression. Epigenetic mechanisms include DNA methylation of promoter and/or non-promoter CpG islands, covalent histone modifications (methylation, acetylation, phosphorylation, ubiquitination or sumoylation), microRNAs, and the more recently described long noncoding RNAs [95, 105–107].
DNA methylation represents a major epigenetic regulatory pathway that is catalyzed by DNA methyltransferases (DNMTs). These enzymes add methyl groups from S-adenosylmethionines to carbon 5 positions of cytosines [108]. Three important family members of DNMTs have been reported: DNMT1, DNMT3A, and DNMT3B. DNMT1 is considered the maintenance DNA methyltransferase, whereas DNMT3A and DNMT3B are primarily involved in de novo DNA methylation [95, 109]. Covalent histone modifications, including acetylation and methylation, are a second major pathway. Histone acetylation and deacetylation are regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively. Histone methylation and demethylation are regulated by histone methyltransferases (HMTs) and histone demethylases (HDMs), respectively [110]. Lysine residues on histone tails can be regulated by both methylation and acetylation. Modifications of lysines on histone H3 can be repressive (e.g., H3K9 di- or trimethylation, H3K27 trimethylation) or activating (e.g., H3K9 acetylation, H3K4 trimethylation) [95, 110–112]. HATs do not bind to DNA promoters directly, but are recruited by DNA-bound transcription factors. Overall, hypomethylation of DNA and histone acetylation cause a more relaxed chromatin state, allowing easier interaction between the DNA and the transcriptional machinery, thus resulting in increased gene transcription. DNA hypermethylation and histone deacetylation result in condensed chromatin structure, causing a decrease in gene transcription [95].
The epigenome is particularly susceptible to environmental factors during fetal development as the DNA synthetic rate is high and the complex DNA methylation patterning and chromatin structure required for normal tissue development is established at this time [113]. DNA methylation and histone modifying pathways cross-talk and both are necessary for normal genomic imprinting during development [114]. Epigenetic reprogramming involves genome-wide erasure and re-establishment of DNA methylation and histone modifications during normal mammalian development (immediately after fertilization until pre-implantation of the embryo and also during primordial germ cell development) [109, 111, 115–118]. Aberrant epigenetic regulation is also implicated in cancer, whereby oncogenes are expressed, and tumor suppressor genes are silenced [112, 119].
In 1991, Garro et al. showed that administration of alcohol to pregnant mice on days 9–11 of pregnancy resulted in genome-wide hypomethylation in 11-day old fetuses. The effect was proposed to be mediated by acetaldehyde, a product of alcohol metabolism, which inhibits DNA methyltransferase in vitro [120]. In the last few years numerous studies have confirmed that alcohol exposure in utero induces epigenetic alterations in the offspring. Studies with the Agouti mouse model demonstrate that alcohol exposure during gestation can affect adult phenotype through epigenetic mechanisms [121]. Microarray analysis of murine embryos exposed to alcohol on gestational day 9 shows altered expression of a subset of genes involved in methylation and chromatin remodeling [122]. In support of this finding, analysis of whole-embryo murine cultures treated with alcohol show increased and decreased DNA methylation of 1,028 and 1,136 genes, respectively. Greater than 200 of these methylation alterations are found on chromosomes 7, 10, and X, which are chromosomes that contain many imprinted genes as well as genes prone to aberrant epigenetic silencing. Additionally, changes in DNA methylation correspond with actual changes in gene expression in 84 genes, indicating that fetal alcohol can impact gene expression in the developing embryo through an epigenetic mechanism [123].
In addition to global methylation analyses, several studies have focused on methylation changes in specific genes. Exposure of mice to alcohol between gestational days 10 and 18 affects methylation of the paternally imprinted H19 gene in the offspring’s sperm, resulting in a 3 % decrease in the number of methylated CpGs in this gene at 8 weeks of age. The CCCTC-factor DNA-binding sites of H19 play a role in regulating IGF-II expression [124]. Due to the well-defined neurological consequences of developmental alcohol exposure, recent studies have focused on epigenetic regulators expressed specifically in the brain. Perinatal alcohol exposure (gestational day 1 through PND 10) induces gene expression changes in epigenetic regulators (DNMT1, DNMT3a, and methyl-CpG binding protein 2) in the hippocampus as well as increases in DNMT activity [125]. Alcohol exposure of fetal cerebral cortical neuroepithelial stem cells causes a decrease in trimethylation of H3K4 (H3K4me3) and H3K27 (H3K27me3) in developmental genes that play an integral role in regulating neural stem cells as well as neural differentiation [126]. H3K4me3 is usually associated with gene activation, whereas H3K27me3 is considered a repressive histone mark [95, 126]. Expression of epigenetic-modifying enzymes such as Dnmt1, Uhrf1, Ash2L1, Wdr5, Ehmt1, and Kdm1b are also observed [126]. DNMT expression and histone modifications are also altered in the hypothalamus of offspring exposed to fetal alcohol. In utero alcohol exposure from gestational day 7–21 results in a decrease in H3K4 di- and trimethylation as well as H3K9 acetylation, and an increase in H3K9 dimethylation as well as expression of DNMT1, DNMT3a, and methyl-CpG-binding protein [127]. These studies raise the possibility that alcohol exposure in utero will affect epigenetic regulators in other tissues as well, including the ovaries and mammary gland.
Related posts:
 A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
 Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
 Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
 Alcohol and HCV: Implications for Liver Cancer
Alcohol and HCV: Implications for Liver Cancer
 Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


