Abbreviations
- ACTH Adrenocorticotropic hormone
- AFC Antral follicle count
- AIRE Autoimmune regulator gene
- AIS Androgen insensitivity syndrome
- AMH Anti-Müllerian hormone
- APS Autoimmune polyglandular syndrome
- ArKO Aromatase knockout
- ART Assisted reproductive therapy
- BBT Basal body temperature
- BMD Bone mineral density
- BMP Bone morphogenic protein
- cAMP Cyclic adenosine monophosphate
- CEE Conjugated equine estrogen
- CRH Corticotropin-releasing hormone
- DHEAS Dehydroepiandrosterone sulfate
- DHT Dihydrotestosterone
- DXA Dual-energy x-ray absorptiometry
- ERA Estrogen Replacement and Atherosclerosis Trial
- FGFR1 Fibroblast growth factor receptor 1
- FMR1 Fragile X gene
- FSH Follicle-stimulating hormone
- FXTAS Fragile-X tremor ataxia syndrome
- GH Growth hormone
- GnRH Gonadotropin-releasing hormone
- hCG Human chorionic gonadotropin
- HDL High-density lipoprotein
- HERS Heart and Estrogen-Progestin Replacement Study
- HPO Hypothalamic pituitary ovarian
- HRT Hormone replacement therapy
- HSD Hydroxysteroid dehydrogenase
- HT Hormone therapy
- IGF-BP Insulin-like growth factor binding protein
- IUI Intrauterine insemination
- LDL Low-density lipoprotein
- LH Luteinizing hormone
- MBH Medial basal hypothalamus
- MCR Metabolic clearance rate
- MMP Matrix metalloproteinase
- MPA Medroxyprogesterone acetate
- MT Menopausal transition
- OMI Oocyte maturation inhibitor
- PCOS Polycystic ovarian syndrome
- POF Premature ovarian failure
- PR Production rate
- PRL Prolactin
- SERM Selective estrogen receptor modulator
- SF-1 Steroidogenic factor-1
- SHBG Sex hormone–binding globulin
- SNRI Serotonin-norepinephrine reuptake inhibitor
- SR Secretion rate
- SRY Sex-determining region of the Y gene
- SSRI Selective serotonin reuptake inhibitor
- StAR Steroidogenic acute regulatory protein
- STRAW Stages of Reproductive Aging Workshop
- SWAN Study of Women’s Health Across the Nation
- Tg Thyroglobulin
- TGF Transforming growth factor
- TPO Thyroid peroxidase
- TRH Thyrotropin-releasing hormone
- TSH Thyroid-stimulating hormone
- VEGF Vascular endothelial growth factor
- WHI Women’s Health Initiative
Embryology and Anatomy
No gene has yet been identified that generates an ovary from an undifferentiated gonad. It is only in the absence of the sex–determining region of the Y gene (SRY) that the gonad develops into an ovary. (For more details, see the discussion of sexual differentiation in Chapter 14.) Primordial germ cells, which give rise to oocytes or spermatogonia, are first identifiable in the yolk sac endoderm (hindgut) at 3 to 4 weeks’ gestation. Once specified, they migrate and proliferate en route through the dorsal mesentery into the gonadal ridge, which is located lateral to the dorsal mesentery of the gut and medial to the mesonephros (Figure 13–1). Studies in mice have suggested that the process of proliferation and navigation to the gonad depends on several genes, including Steel (kit ligand and receptor), β1 integrin, pog (proliferation of germ cells), and many cytokines. Failure of primordial germ cells to develop or migrate into the gonadal ridge results in failure of ovarian development. In contrast, it is suggested that male gonadal development may continue to develop into functional testis despite the absence of germ cells.
Figure 13–1
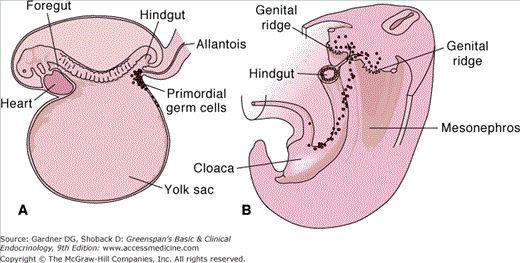
A. Schematic drawing of a 3-week-old embryo showing the primordial germ cells in the wall of the yolk sac, close to the attachment of the allantois. B. Drawing to show the migrational path of the primordial germ cells along the wall of the hindgut and the dorsal mesentery into the genital ridge.
(Reproduced, with permission, from Langman J, Sadler TW. Langman’s Medical Embryology. 8th ed. Lippincott Williams & Wilkins; 2000.)
The germ cells that reach the gonadal ridge (6 weeks’ gestation) continue to proliferate and are referred to as oogonia (premeiotic germ cells). At 10 to 12 weeks’ gestation, some oogonia leave the mitotic pool and begin meiosis, where they arrest in prophase I (dictyotene stage). These arrested germ cells are now called primary oocytes. By 16 weeks, primordial follicles are first identified, making a clear distinction for gonadal differentiation into an ovary. At approximately 20 weeks’ gestation, a peak of 6 to 7 million germ cells (two-thirds of them primary oocytes and one-third oogonia) are present in the ovaries. During the second half of gestation, the rate of mitosis rapidly decreases and the rate of oogonial and follicular atresia increases. Those oogonia that are not transformed into primary oocytes will undergo atresia before birth. This leads to a reduction in the number of germ cells, resulting in a total of 1 to 2 million germ cells at birth. No germ cell mitosis occurs after birth, whereas follicular atresia continues, with the result that the average girl entering puberty has only 300,000 to 400,000 germ cells.
The ovary is organized into an outer cortex and an inner medulla. The germ cells are located within the cortex. Along the outer surface of the cortex is the germinal epithelium. This cell layer is composed of cuboidal cells resting on a basement membrane and forms a continuous layer with the peritoneum. Even though it is called the germinal epithelium, there are no germ cells within this layer. During embryonic development, the epithelial cells proliferate and enter the underlying tissue of the ovary to form cortical cords. When the primordial germ cells arrive at the genital ridge, they are incorporated into these cortical cords. At the same time the germ cells migrate from the yolk sac, the stromal cells of the ovary (granulosa and interstitial cells) migrate from the mesonephric tubules into the gonad. Primordial follicles form within the cortical cords. They are composed of a primary oocyte and one layer of granulosa cells with its basement membrane. Oocytes not surrounded by granulosa cells are lost, probably by apoptosis. This finite follicle population represents the pool of germ cells that will ultimately be available to enter the follicular cycle.
During fetal development, the gonad is held in place by the suspensory ligament at the upper pole and the gubernaculum at the lower pole. The final location of the gonad is dependent on hormone production. In the presence of testosterone, the gubernaculum grows while the suspensory ligament regresses. As the gubernaculum continues to grow, the gonad (testis) descends into the scrotum. In contrast, when testosterone is absent, the suspensory ligament remains and the gubernaculum regresses. This process maintains the gonad (ovary) in the pelvis.
The remainder of the female internal reproductive organs are formed from the paramesonephric (Müllerian) ducts. In the absence of anti-Müllerian hormone (AMH), the paramesonephric system develops into the uterine (fallopian) tubes, uterus, cervix, and upper third of the vagina. Unlike Wolffian duct differentiation, the development of the female reproductive tract is not dependent on hormone production. (For more details, see “Human Sex Differentiation” in Chapter 14.) Briefly, the Müllerian buds are formed lateral to the Wolffian ducts and the gonadal ridge after 37 days’ gestation (Figure 13–2). These buds elongate, extend caudally, and cross medially to the Wolffian ducts by 8 weeks’ gestation. By 10 weeks, the adjacent paired Müllerian ducts meet in the midline as they join the Müllerian tubercle (urogenital sinus). Over the next couple of weeks, the Wolffian ducts degenerate, and the paired Müllerian ducts fuse and begin to canalize. The intervening septum resorbs between the 12th and 16th weeks of gestation, resulting in a single uterine cavity. The most cranial parts of the Müllerian ducts remain unfused and form the uterine tubes, which remain patent with the coelom (future peritoneal cavity). The caudal segments stimulate solid cords to extend from the Müllerian tubercle to the sinovaginal bulbs from the posterior aspect of the urogenital sinus. In turn, the sinovaginal bulbs extend cranially and fuse with the vaginal cords, forming the vaginal plate. The vagina is subsequently formed by canalization of the vaginal plate. It is ultimately the cervix and the upper one-third of the vagina that are derived from the Müllerian structures, and the remaining lower two-thirds are formed from the urogenital sinus.
Figure 13–2
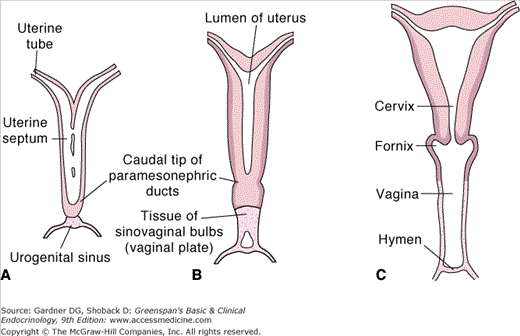
Schematic drawing showing the formation of the uterus and vagina. A. At 9 weeks. Note the disappearance of the uterine septum. B. At the end of the third month. Note the tissue of the sinovaginal bulbs. C. Newborn. The upper portion of the vagina and the fornices are formed by vacuolization of the paramesonephric tissue and the lower portion by vacuolization of the sinovaginal bulbs.
(Reproduced, with permission, from Langman J, Sadler TW. Langman’s Medical Embryology. 8th ed. Lippincott Williams & Wilkins; 2000.)
The uterus is composed of endometrium (innermost lining), myometrium, and serosa. The adult uterus is a pear-shaped hollow organ. The cervical portion extends approximately 2 cm into the vagina, and the remaining corpus extends approximately 6 cm into the abdomen. The normal adult uterus weighs 40 to 80 g.
The uterus is located in the pelvis and rests on the pelvic floor. Seventy percent to 80% of the time, the uterine position is anteflexed (cervical-uterine corpus angle) and anteverted (cervical-vaginal angle). Therefore, when a woman is standing, the corpus of the uterus is horizontal and resting on top of the bladder. The uterus has several paired ligaments that develop from thickenings of the peritoneum and serve to maintain this anatomic position. The cardinal ligament (Mackenrodt) is the main supporting ligament. It attaches to the lateral margins of the cervix at the upper vagina and extends to the lateral pelvic wall. The remaining ligaments—uterosacral, round, and broad—have a lesser role in supporting the uterus.
The uterine artery originates from the anterior division of the internal iliac artery (hypogastric), enters the cardinal ligament, and supplies the uterus. The uterine artery divides into a descending branch and an ascending branch known as the vaginal and arcuate arteries, respectively. The arcuate arteries anastomose with each other and form a vascular network around the uterus. The radial arteries branch off from the arcuate network and penetrate the uterus to supply the myometrium. Smaller basal branches and spiral arteries supply the endometrium.
The ovary is suspended in the pelvis and has three associated ligaments. The average adult ovary is 2.5 to 5 cm × 2.5 cm × 1 cm in size and weighs 3 to 8 g. The position of the ovary is variable, but in a nulliparous woman it is often located in a peritoneal depression on the pelvic sidewalls between the ureter and external iliac vein. The suspensory (infundibulopelvic) ligament attaches to the cranial pole of the ovary and extends to the pelvic brim. This ligament suspends the ovary in the pelvis and contains the ovarian vessels, lymphatics, and nerves. The utero-ovarian ligament attaches to the inferior pole of the ovary and extends to the uterus. The mesovarium connects the anterior portion of the ovary to the posterior leaf of the broad ligament. The blood supply of the ovary originates from the abdominal aorta, passes through the suspensory ligament, and enters the mesovarium to form an anastomotic network with branches from the uterine artery. The ovarian artery enters the ovarian hilum and branches into spiral arteries that enter the medulla and extend to the ovarian cortex. Other branches from the anastomotic network, located in the mesovarium, supply the uterine tubes.
Ovarian Steroidogenesis
The ovaries are not only the store for germ cells—they also produce and secrete hormones that are vital for reproduction and the development of secondary sexual characteristics. The next section will briefly discuss the biosynthesis of ovarian hormones.
In the ovary, the major source of hormone production is the maturing follicle. The components of the follicle are the theca cells, the granulosa cells, and the primary oocyte. The theca cells produce androgens, and the granulosa cells produce estrogens. The other stromal cells that contribute to androgen production can be divided into two populations of cells: the secondary interstitial cells (derived from theca) and the hilum cells. These cells are the major ones involved in ovarian hormone production during menopause (see below).
The ovarian hormones are derived from cholesterol. Steroidogenic cells acquire the cholesterol substrate from one of three sources. The most common source is plasma lipoprotein-carrying cholesterol, primarily in the form of low-density lipoprotein (LDL). Other minor sources include de novo synthesis from acetate and liberation from stored lipid droplets (cholesterol esters). Stimulation of ovarian cells by trophic hormones such as follicle-stimulating hormone (FSH) and luteinizing hormone (LH) facilitate uptake of cholesterol by increasing the number of LDL receptors on the cell surface. The LDL particle is subsequently internalized and degraded in the lysosome. The free cholesterol that is liberated from the lysosome is delivered to the mitochondria by an unknown mechanism, possibly via microfilaments and microtubules. The cholesterol is then translocated into the mitochondria by the steroidogenic acute regulatory protein (StAR). Thus, StAR is the rate-limiting step regulating substrate availability for steroidogenesis.
Although acute alterations in steroid production result from changes in delivery of cholesterol to the mitochondria, the long-term control of steroid synthesis results from regulation of gene expression. Most of the genes involved in steroidogenesis contain at least one steroidogenic factor-1 (SF-1) response element in the promoter region. These elements are critical for the regulation of steroidogenic genes as well as the development of adrenal gland, ovary, and testis. The importance of SF-1 for steroidogenesis is highlighted in knockout mice deficient in this transcription factor which lack adrenal glands and gonads. Although SF-1 is essential, the specific expression of each gene involves many other transcription factors that work independently or in concert with SF-1.
The rate-determining step that commits cholesterol to steroid synthesis is the cholesterol side chain cleavage enzyme reaction (P450scc) (Figure 13–3). This reaction converts cholesterol to pregnenolone, the precursor of steroid hormones, and takes place in the mitochondria. Pregnenolone is transported out of the mitochondrion, and the remaining steps in sex steroid production take place primarily within the smooth endoplasmic reticulum (SER).
Figure 13–3
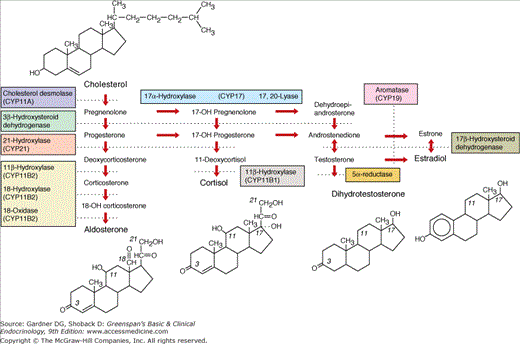
Pathways of steroid biosynthesis. The pathways for synthesis of progesterone and mineralocorticoids (aldosterone), glucocorticoids (cortisol), androgens (testosterone and dihydrotestosterone), and estrogens (estradiol) are arranged left to right. The enzymatic activities catalyzing each bioconversion are written in the boxes. For those activities mediated by specific cytochrome P450, the systematic name of the enzyme (CYP followed by a number) is listed in parentheses. CYPB2 and CYP17 have multiple activities. The planar structures of cholesterol, aldosterone, cortisol, dihydrotestosterone, and estradiol are placed near the corresponding labels.
(Reproduced, with permission, from White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000;21:245.)
Once pregnenolone is formed, the particular hormones that are synthesized are dependent on the endocrine organ and cell type. For example, the main sources of sex steroids in the female come from the adrenal gland, ovary, and the periphery. The specific type of hormone synthesized is dependent on the specific gene expression within each cell type.
In the adrenal gland, there are three zones: zona glomerulosa, zona fasciculata, and zona reticularis. The cells in the different zones start with the same hormone precursor but differ in their secretory products. The glomerulosa produces mainly aldosterone, whereas cortisol and androgen are produced by the zona fasciculata and zona reticularis, respectively. The major androgen produced by the adrenal gland is dehydroepiandrosterone sulfate (DHEAS). Differences in enzymatic activity among cells in the various zones are what regulate hormone production. The zona reticularis and zona fasciculata lack 11β-hydroxylase, which is necessary for aldosterone synthesis (see Figure 13–3; see Chapters 9 and 10). The zona glomerulosa lacks 17-hydroxylase and 17,20-lyase (CYP17), which are necessary for sex steroid synthesis.
Ovarian cells similarly secrete different hormones due to differential enzyme activity. The theca interstitial and secondary interstitial cells lack aromatase and hence are the androgen producers in the ovarian cortex. The granulosa cells, on the other hand, lack CYP17 and, therefore, secrete estrogens—mainly estradiol—from thecal cell-derived androgens in the proliferative phase and progesterone in the luteal phase (see later).
Adipose and skin make significant contributions to plasma concentrations of some sex steroids. Adipose tissue is able to sequester most steroids due to their lipid solubility. Fat also expresses genes capable of sex steroid metabolism (ie, aromatase). Skin significantly contributes to the plasma concentration of testosterone by utilizing circulating DHEAS and androstenedione as precursors.
The major circulating androgens include DHEAS, androstenedione, and testosterone. During the reproductive years, the ovaries are directly responsible for one-third of the testosterone production. The remaining two-thirds come from the periphery (17β-hydroxysteroid dehydrogenase [HSD] types 3 and 5) and is derived from ovarian and adrenal gland precursors—notably androstenedione, which is produced in equal proportions by the adrenal gland and the ovary. The adrenal gland may directly secrete testosterone, but its major contribution is derived from its production of precursors. Therefore, the ovaries are responsible for nearly two-thirds of the circulating testosterone. This differs from males, in whom only 5% of the circulating testosterone is derived from peripheral conversion of androstenedione. It is also estimated that more than 60% of the most potent androgen, dihydrotestosterone (DHT) in women, is produced in the skin (5α-reductase type 1 and 2) and originates from androstenedione. DHEAS is the major androgen produced by the adrenal gland. The adrenal gland is responsible for more than 95% of the circulating DHEAS levels. Although it is the most abundant androgen circulating in the body, it contributes minimally to serum testosterone levels. The relative strengths of androgens are listed in Table 13–1.
The circulating estrogens include estrone, estrone sulfate, estradiol, and estriol (pregnancy). More than 95% of estradiol in the circulation is produced from the ovary. In contrast to estradiol, approximately one-half of the circulating estrone is secreted from the ovary, while the remaining is derived from peripheral conversion. The most significant precursor, androstenedione, is aromatized in the adipose tissue, hair follicles, and the liver to estrone. Estrone is also derived from estradiol through 17β-HSD type 2 activity or from estrone sulfate (steroid sulfatase). Almost all estriol is produced during pregnancy and is secreted from the placenta (see Chapter 16).
Ultimately, the serum concentration of sex steroids is dictated by the secretion rate (SR), production rate (PR), and metabolic clearance rate (MCR), as shown in Table 13–2. The SR of sex steroids from each organ determines the PR of the hormone. If the SR for a specific hormone in the ovary equals the PR, then there is no extragonadal formation. However, if there is extra-gonadal formation, then the PR exceeds the SR.
| Concentration in Plasma | ||||||
|---|---|---|---|---|---|---|
| Compound | MCR of Compound in Peripheral Plasma (L/d) | Phase of Menstrual Cycle | (nmol/L) | (μg/dL) | PR of Circulating Compound (mg/d) | SR by Both Ovaries (mg/d) |
| Estradiol | 1350 | Early follicular Late follicular Midluteal | 0.2 1.2–2.6 0.7 | 0.006 0.033-0.070 0.020 | 0.081 0.445-0.945 0.270 | 0.07 0.4-0.8 0.25 |
| Estrone | 2210 | Early follicular Late follicular Midluteal | 0.18 0.5-1.1 0.4 | 0.005 0.015-0.030 0.011 | 0.110 0.331-0.662 0.243 | 0.08 0.25-0.50 0.16 |
| Progesterone | 2200 | Follicular Luteal | 3.0 36 | 0.095 1.13 | 2.1 25.0 | 1.5 24.0 |
| 20α-Hydroxyprogesterone | 2300 | Follicular Luteal | 1.5 7.5 | 0.05 0.25 | 1.1 5.8 | 0.8 3.3 |
| 17-Hydroxyprogesterone | 2000 | Early follicular Late follicular Midluteal | 0.9 6 6 | 0.03 0.20 0.20 | 0.6 4.0 4.0 | 0-0.3 3-4 3-4 |
| Androstenedione | 2010 | 5.6 | 0.159 | 3.2 | 0.8-1.6 | |
| Testosterone | 690 | 1.3 | 0.038 | 0.26 | ||
| Dehydroepiandrosterone | 1640 | 17 | 0.490 | 8.0 | 0.3-3 | |
MCR is the volume of blood per unit of time cleared of the hormone. The metabolic clearance rate of sex steroids is inversely related to the affinity for sex hormone–binding globulin (SHBG) and/or albumin. Prior to excretion, steroids are conjugated to make them water soluble. The bulk of testosterone is bound to SHBG (∼65%) and to a lesser extent albumin (∼35%); only 1% (free hormone) is active and available for metabolism. The remaining androgens have negligible binding affinity for SHBG. Free testosterone may be converted to more potent androgens such as DHT or may be metabolized through androstenedione. Metabolites of androstenedione and DHT are conjugated with a sulfate or glucuronide and are excreted in the urine. The majority of estradiol is also bound, although it has less binding affinity than testosterone for SHBG (38%) and more for albumin (60%); approximately 2% is unbound. Estradiol may be directly conjugated (16 α-hydroxylated or 2-hydroxylated) or is metabolized to estrone prior to conjugation. The remaining estrogens are weakly bound to proteins. Progesterone is metabolized into many intermediates prior to conjugation. Pregnanediol glucuronide is the major metabolite observed in the urine.
Physiology of Folliculogenesis and the Menstrual Cycle
The menstrual cycle is regulated by complex interactions between the hypothalamic-pituitary-ovarian (HPO) axis and the uterus. Briefly, the hypothalamus secretes gonadotropin-releasing hormone (GnRH), which stimulates the pituitary to release FSH and LH. These gonadotropins then trigger the ovary to release an oocyte that is capable of fertilization. Concurrently, the ovary secretes hormones, which act on the endometrial lining of the uterus to prepare for implantation. In addition, the ovarian hormones feed back to the hypothalamus and pituitary, regulating the secretion of gonadotropins during the phases of the menstrual cycle. This complex interaction will be discussed in greater detail below. The hormonal changes associated with menstruation are summarized in Figure 13–4.
Figure 13–4
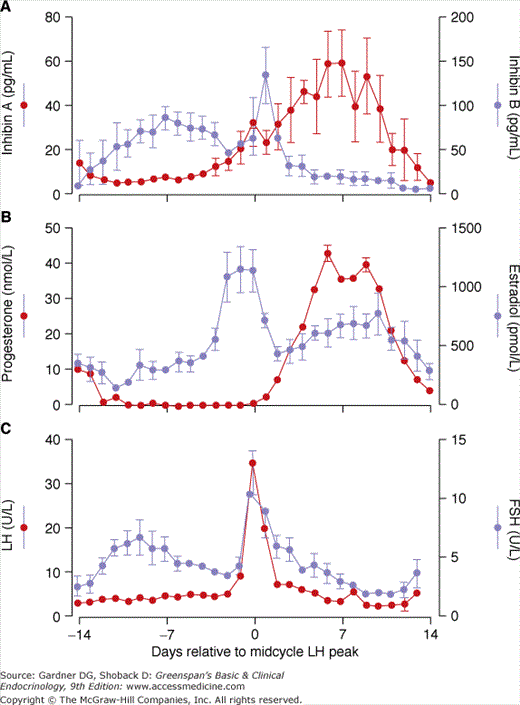
The endocrinology of the luteal-follicular transition in women. Data are mean ± SE of daily serum concentrations of FSH, LH, estradiol, progesterone, and immunoreactive inhibin in women with normal cycles. Note the secondary rise in plasma FSH in the late luteal phase (∼2 days before menses).
(Reproduced with permission from Erickson GF. Ovarian anatomy and physiology. In: Lobo R, ed. Menopause: Biology and Pathobiology. Academic Press; 2000.)
GnRH is the central initiator of reproduction. GnRH is a 10 amino acid peptide with a short half-life of 2 to 4 minutes. It is processed in specialized secreting neurons that originate in the olfactory placode during development and migrate to the arcuate nucleus of the medial basal hypothalamus (MBH). These neurons project from the median eminence and secrete GnRH, with inherent rhythmic behavior (pulse generator), into the portal vessels to reach the gonadotropes within the anterior pituitary. GnRH binds to its receptor, a member of the G protein–coupled seven-transmembrane-spanning receptor superfamily. 1,4,5–inositol triphosphate and diacylglycerol act as second messengers for GnRH. The pulsatile frequency of GnRH secretion regulates gonadotropin synthesis and the secretion of pituitary gonadotropes (see Chapter 4).
During the late luteal-follicular phase, the slower pulsatile release of GnRH—every 90 to 120 minutes—favors FSH secretion. In response to FSH, the maturing follicle in the ovary secretes estradiol. This hormone is involved in a negative feedback loop that inhibits the release of FSH by indirectly decreasing the production of GnRH via gamma-aminobutyric acid neurons, in addition to possibly having a direct effect on the pituitary gland. Estradiol is also involved in a positive feedback loop that increases the frequency of GnRH pulses to every 60 minutes during the follicular phase and acts directly on the pituitary to stimulate LH secretion. LH stimulates the ovary to further increase estradiol production (see two-cell theory, later). There is no further change in GnRH pulsatility at this point in the cycle; however, estradiol and other regulatory factors (see later) increase pituitary sensitivity to GnRH. This increased sensitivity results in a rapid elevation of LH production—the LH surge—which stimulates ovulation. After ovulation, the ruptured follicle (corpus luteum) secretes progesterone. This hormone is involved in a negative-feedback loop, indirectly through increased endogenous opioid activity and possibly directly through a reduction in GnRH pulsatility to every 3 to 5 hours. This favors FSH synthesis during the luteal-follicular transition. As progesterone levels fall again, GnRH pulsatility increases, favoring FSH release.
Gonadotropes are located in the adenohypophysis and make up approximately 10% of the cells in the pituitary. These cells synthesize and secrete FSH and LH. These hormones, thyroid-stimulating hormone (TSH) and human chorionic gonadotropin (hCG), all belong to a family of glycoprotein hormones. Gonadotropins are functional as heterodimers and are composed of an alpha and a beta subunit. The alpha subunit amino acid sequence is identical for all of the glycoprotein hormones while the beta subunits contain different amino acid sequence and confer unique specificity on the glycoproteins.
The synthesis of FSH and LH mostly occur in the same cell, yet their secretion patterns differ. The secretion of FSH is tightly linked to the expression of the FSH beta subunit. This suggests that there is minimal storage of FSH within the gonadotropes, and the majority of secretion follows more of a constitutive pathway. This is in contrast to LH secretion, in which it is first stored in organelles and then released with the appropriate trigger (regulated pathway). The different oligosaccharides on the beta subunits likely facilitate the intracellular sorting that results in different mechanisms of secretion.
The differential gene expression that leads to the production and release of gonadotropins by cells in the pituitary is influenced by GnRH and ovarian hormones through feedback loops. Slower GnRH pulse frequency enhances FSH beta subunit expression and increases LH amplitude. In turn, increased GnRH pulse frequency stimulates LH beta subunit expression while promoting FSH release. As a result, LH amplitude decreases while the mean concentration rises. Thus, ovarian steroid modification of hypothalamic GnRH pulsatility controls pituitary gonadotrophin production.
An intrapituitary network involves several factors that play a role in regulating gonadotropin synthesis and secretion. The gonadotropes produce and secrete peptides that are in the transforming growth factor (TGF) family. Activin is a local regulatory protein that is involved in control of gonadotrope function. Slow pulses of GnRH enhance activin synthesis, which subsequently enhances FSH transcription. Follistatin, another TGF-related protein that binds to activin, is stimulated by rapid pulses of GnRH. This decreases the bioavailability of activin and consequently reduces FSH synthesis. In addition to these local modifiers, ovarian transforming growth factors such as inhibin also modulate the expression of gonadotropins (see later).
The ovary is intimately involved in regulating the menstrual cycle via steroid feedback to alter gonadotropin secretion. In addition, the ovary contains an intraovarian network involving factors that are synthesized locally and have a paracrine and autocrine role in the modulation of gonadotropin activity. The intraovarian regulators include the insulin-like growth factor (IGF) family, TGF superfamily, and epidermal growth factor (EGF) family. Furthermore, it is these factors that assist in the coordination of follicular development, steroidogenesis, and ovulation.
The menstrual cycle of the ovary includes a follicular phase and a luteal phase. The follicular phase is characterized by growth of the dominant follicle and ovulation. It typically lasts 10 to 14 days. It is, however, this phase that is variable in duration and most often accounts for the variability in menstrual cycle length in ovulatory women. The luteal phase starts after ovulation and is the period when the ovary secretes hormones that are essential to accommodate conceptus implantation. This phase is relatively constant and averages 14 days (range, 12-15 days) in duration. The next section will describe the two phases in some detail.
Primordial follicles are the fundamental reproductive units that comprise the pool of resting oocytes. Morphologically, they are composed of a primary oocyte that is surrounded by a single layer of squamous granulosa cells and a basement membrane. They have no blood supply. These primordial follicles develop between the sixth and ninth months of gestation and harbor the complete supply of ovarian follicles.
The initiation of follicular growth begins with the transition of the dormant primordial follicle into the growth phase. The exact mechanisms controlling the initial recruitment of the primordial follicle are under investigation. It is suggested that the resting follicular pool is probably under tonic inhibitory control. The initial recruitment process induces growth in some primordial follicles; other neighboring follicles remain quiescent for many months or years. It is thought that the recruitment of these follicles is a continuous process that begins once the germ pool is created and ends with follicular exhaustion. This complex process is gonadotropin independent. Several studies have suggested that an intraovarian signaling network, involving members of the TGF superfamily, is responsible for primordial follicle recruitment. It is also known that recruitment and growth of the follicle absolutely requires cell-to-cell contact with neighboring granulosa cells and the oocyte. These cells transfer various factors, nutrients, and waste to and from the oocyte through gap junctions derived from connexins.
One theory proposes that the oocyte actively influences its own fate by secreting various factors. These include two particular growth factors related to TGF-β, which are produced by the oocyte early in follicular development, growth differentiation factor (GDF)-9 and bone morphogenic protein (BMP)-15. Knockout mouse studies suggest that the oocyte induces granulosa cell proliferation through these growth factors, and the granulosa cell responds with factors (eg, follistatin, c-kit) to decrease the inhibitory influences (eg, activin A, Müllerian-inhibiting substance) and promote stimulators of oocyte growth (Figure 13–5).
Figure 13–5
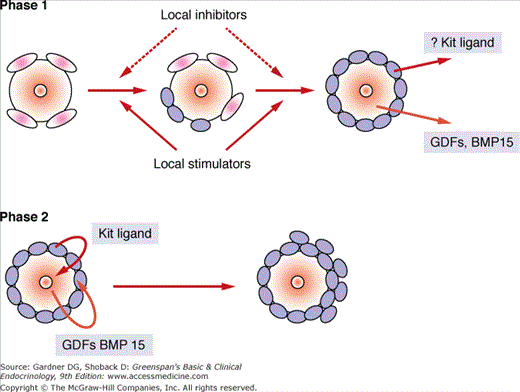
A theoretical model of follicular recruitment into the growth phase. Initiation of follicular growth may be regulated by many local inhibitory and stimulatory factors (ie, inhibin A, follistatin, kit ligand) and begins with transformation of the granulosa cells from squamous to cuboidal shape. Oocytes subsequently commence growth and regulate proliferation and differentiation of granulosa cells with members of the transforming growth factor β superfamily (ie, GDF-9, BMP15). Phase 1: transition of granulosa cells from flattened to cuboidal. Phase 2: oocyte growth and proliferation and differentiation of granulosa cells.
(Redrawn with permission of Braw-Tal R. The initiation of follicle growth: the oocyte of the somatic cells? Mol Cell Endocrinol. 2002;187:11.)
Several other local factors have been described to date, and many more will be identified in the future. The ongoing search for these growth factors and hormones will ultimately elucidate the physiology of primordial follicle recruitment. There is a finite number of germ cells, and each successive recruitment further depletes the germ pool. Any abnormality that alters the number of germ cells or accelerates recruitment could perhaps lead to early ovarian follicular depletion and, therefore, early reproductive failure (see section on infertility, later).
Primary follicle development is the first stage of follicular growth (Figure 13–6). Primary follicles differ from primordial follicles in several ways. The oocyte begins to grow. As growth progresses, the zona pellucida is formed. This is a thick layer of glycoprotein that is most likely synthesized by the oocyte. It completely surrounds the oocyte and forms a barrier between the oocyte and the granulosa cell layer. It serves a number of biologic functions that are critical for protection of the oocyte and conception. Finally, the granulosa cells undergo a morphologic change from squamous to cuboidal. This stage of development may last 150 days.
Figure 13–6
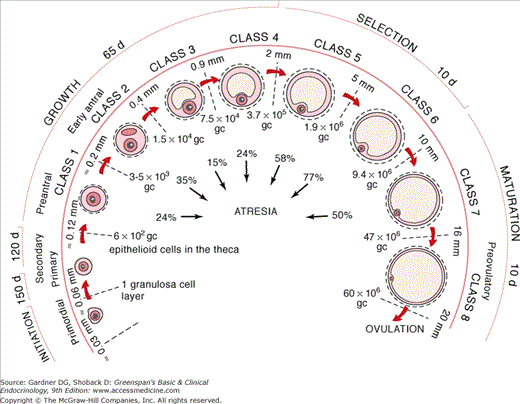
The chronology of folliculogenesis in the human ovary. Folliculogenesis is divided into two major periods, preantral (gonadotropin independent) and antral (FSH dependent). In the preantral period, a recruited primordial follicle develops into the primary/secondary (class 1) and early tertiary (class 2) stages, at which time cavitation or antrum formation begins. The antral period includes the small graafian (0.9-5 mm, classes 4 and 5), medium graafian (6-10 mm, class 6), large graafian (10-15 mm, class 7), and preovulatory (16-20 mm, class 8) follicles. Time required for completion of preantral and antral periods is approximately 300 and 40 days, respectively. Number of granulosa cells (gc), follicle diameter (mm), and atresia (%) are indicated.
(Reproduced, with permission, from Gougeon A. Regulation of ovarian follicular development in primates: facts and hypotheses. Endocr Rev. 1996;17:121.)
The progression to a secondary follicle includes attainment of maximal oocyte growth (120 μm in diameter), proliferation of granulosa cells, and acquisition of theca cells. The exact mechanism involved in acquiring theca cells is not completely understood, but they are thought to be derived from the surrounding ovarian mesenchyme (stromal fibroblasts) as the developing follicle migrates to the medulla. It is the development of this layer that gives rise to the theca interna and theca externa. With theca cell development, these follicles gain an independent blood supply, although the granulosa cell layer remains avascular. In addition, the granulosa cells in the secondary follicle develop FSH, estrogen, and androgen receptors. This phase of follicular development may take as long as 120 days, probably because of the long doubling time (>250 hours) of granulosa cells.
Further follicular development leads to the tertiary follicle or early antral phase. This phase is characterized by the formation of an antrum or cavity in the follicle. The antral fluid contains steroids, proteins, electrolytes, proteoglycans, and an ultrafiltrate that forms from diffusion through the basal lamina. Other changes in this phase include further theca cell differentiation. Subpopulations of thecal interstitial cells develop within the theca interna, acquire LH receptors, and are capable of steroidogenesis. The granulosa cells begin to differentiate into distinct cell layers. Starting from the basal lamina, the cells can be stratified into the membrana, periantral, cumulus oophorus, and corona radiata layers. This developmental process is influenced by FSH and unidentified signals originating from the oocyte. It is suggested that oocyte-derived GDF-9, is an important factor in this process, where the relative concentration of GDF-9 dictates the specific subtype of granulosa cell. In addition, the granulosa cell—most likely in response to FSH—starts producing activin, a member of the TGF family. Activin is composed of two types of beta subunits, βA and βB, which are held together by disulfide bonds. It is the combination of these subunits that generates the various activins (activin A [βA,βA], AB [βA,βB], or BB [βB,βB]). It is unlikely that activin has an endocrine role, because serum activin levels do not change throughout the menstrual cycle and the free activin level in the serum is negligible. Activin’s primary activity is within the ovary, where it plays an autocrine role by enhancing FSH receptor gene expression in the granulosa cells and accelerating folliculogenesis.
Follicular growth during the early antral phase occurs at a slow and constant pace. The follicle achieves a diameter of 400 μm. FSH-stimulated mitosis of granulosa cells is the major contributor to follicular growth at this stage. Until this point, follicular growth and survival are largely independent of gonadotropins. In fact, prepubertal females and women taking oral contraceptives may have follicles arrested at various phases up until this point. It is at this phase in follicular development that FSH is critical for growth and survival. If FSH does not rescue these follicles, they undergo atresia.
The morphologic follicular unit, consisting of theca cells and granulosa cells, is also a functional hormonal unit capable of substantial estrogen production (two-cell theory; Figure 13–7). The thecal interstitial cells and granulosa cells are under the direct influence of LH and FSH, respectively. The gonadotropins increase cyclic adenosine monophosphate (cAMP) production and the activity of the transcription factor SF-1 in the respective cell types.
Figure 13–7
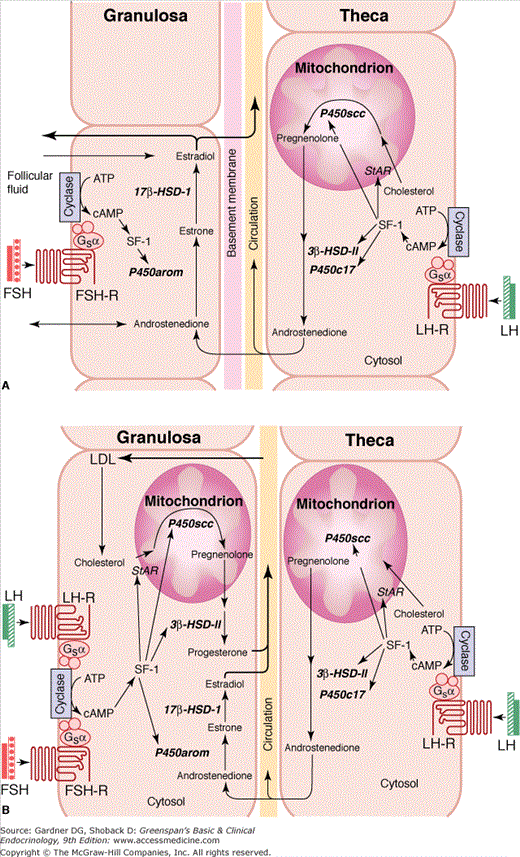
Two-cell hypothesis. A. The preovulatory follicle produces estradiol through a paracrine interaction between theca and granulosa cells. The theca cells provide the substrate, androstenedione, to the granulosa cell for estradiol production. The granulosa cells express the enzymes necessary for transformation of androstenedione to estradiol (P450 aromatase, 17β hydroxysteroid dehydrogenase-1). B. In the corpus luteum, granulosa-lutein cells gain vascularity, LH receptors, and the enzymes necessary for progesterone synthesis. The theca-lutein cells remain the source of androstenedione for estradiol production in granulosa-lutein cells. Gonadotropins, steroidogenic factor-1 (SF-1), and steroidogenic acute regulatory protein (StAR) play crucial roles in steroidogenesis with the ovary (ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; FSH-R, follicle–stimulating hormone receptor; LH-R, luteinizing hormone receptor; P450 arom, P450 aromatase;17β- HSD-I, 17-β hydroxysteroid dehydrogenase-I).
(Redrawn with permission from Larsen et al, eds. The physiology and pathology of the female reproductive axis. In: Williams Textbook of Endocrinology. Saunders; 2003; 604.)
In the theca cell, LH increases the number of LH receptors on the cell surface and the expression and activity of StAR, P450scc, 3β-HSD-II, and P450c17 to increase androgen production. FSH increases the expression of aromatase and 17β-HSD within the granulosa cell.
The pathway of androgen synthesis is shown in Figure 13–3. Androgens may be formed in one of two ways: (1) Δ5 pathway, which entails dehydroepiandrosterone (DHEA) as the precursor to androgens, and (2) Δ4 pathway, which synthesizes androgen through 17-OH progesterone. However, in the human, the contribution of the Δ4 pathway for androgen production appears minimal. This is due to 17,20-lyase activity having a much higher affinity for 17-OH pregnenolone than 17-OH progesterone. Therefore, the main precursor to sex steroids in the human is DHEA.
The androgens—mainly androstenedione—then diffuse through the basal lamina of the follicle and are the main precursors that granulosa cells utilize for estrogen production through the activity of the aromatase enzyme. However, the pathway of estradiol biosynthesis is dictated by the type of 17β-HSD that is expressed. In humans, seven 17β-HSDs have been identified, each having different affinities for particular steroids. In the granulosa cell, it is type 1 17β-HSD that is expressed preferentially reducing estrone to yield estradiol. Type 3 17β-HSD preferentially reduces androstenedione to testosterone and is expressed in the Leydig cells and not in the ovary. However, type 5 17β-HSD is expressed in the theca cells and is likely the catalyst for the final step in testosterone synthesis from androstenedione in the ovary. In summary, the major pathway of estradiol biosynthesis within the granulosa cell entails androstenedione aromatization to estrone by aromatase, followed with a reduction to estradiol by type 1 17β-HSD.
The importance of estradiol on folliculogenesis with respect to its negative and positive feedback effects on gonadotropin secretion is well established. However, the role of estradiol in local oocyte maturation and follicular growth remains controversial. There is evidence that estrogen is synergistic with FSH during the follicular phase by increasing FSH and LH receptors, stimulating proliferation of granulosa cells and gap junctions as well as aromatase activity. The aromatase knockout (ArKO) mouse suggests a local role for estrogen. The ArKO mouse initially has large antral follicles, but after the first year, there are no remaining antral follicles or secondary follicles and atresia is evident among the remaining primary follicles. However, the ArKO oocytes are capable of in vitro maturation and blastocyst formation. There is evidence that both estrogen receptors are expressed in the granulosa and theca cells. Studies with the estrogen receptor α knockout mice show these mice are infertile and lack graafian follicles. However, the estrogen receptor β knockouts are fertile but produce smaller litters. In humans, there are cases where follicular development has occurred in the absence of estradiol secretion. This was observed in a woman with CYP17α deficiency where promotion of follicular growth was noted with gonadotropins. Embryo development followed in vitro fertilization, but unfortunately no pregnancy resulted.
Intraovarian factors play a major modulatory role in both folliculogenesis and steroidogenesis. The oocyte-derived factor GDF-9 is expressed throughout folliculogenesis. It is thought not only to promote granulosa cell differentiation but also to have a stimulatory effect on theca cells and an inhibitory effect on luteal cell formation. The IGFs enhance the response to FSH. In vitro studies have shown that both IGF-1 and IGF-2 increase granulosa cell proliferation and estradiol secretion. However, it is suggested that IGF-2 rather than IGF-1 plays a dominant role in follicular maturation. This may be explained by the absence of IGF-1 expression in the granulosa cells of dominant follicles. Furthermore, women with Laron syndrome (IGF-1 deficiency) can be induced to ovulate by ovarian stimulation with gonadotropins. This scenario suggests that IGF-1 is not critical for folliculogenesis.
The granulosa cells produce other hormones (members of the TGF-β family), that regulate folliculogenesis (see activin earlier). The granulosa cells synthesize the alpha subunit, which combines with beta subunits to create a heterodimer known as inhibin A (αβA) or inhibin B (αβB). The role of inhibins in folliculogenesis and steroidogenesis is largely indirect; it suppresses pituitary FSH secretion. The serum concentrations of inhibin A and B are influenced by the menstrual rhythm. The circulating levels of inhibin A rise late in the follicular phase and remain elevated in the luteal phase whereas the concentration of inhibin B mirrors the menstrual pattern of serum FSH levels (see Figure 13–4). Although inhibin B expression is increased within the granulosa cell on stimulation with FSH, the follicular concentration of inhibin B does not change with follicular size. It has been suggested that the serum concentration of inhibin B reflects the volume of granulosa cells within the ovary, thereby serving as an indicator of the size of the growing cohort of follicles (ovarian reserve). Because inhibin B is the primary inhibitor of pituitary production of FSH in the follicular phase in the absence of estradiol, measuring baseline FSH circulating levels early in the follicular phase (cycle day 3) serves as indirect marker of ovarian reserve (see infertility).
The antral growth phase of follicular development is characterized by rapid follicle growth (1-2 mm/d) and is gonadotropin dependent. In response to FSH, the antral follicle rapidly grows to a diameter of 20 mm, primarily as a result of accumulation of antral fluid. The theca interna continues to differentiate into interstitial cells that produce increasing amounts of androstenedione for aromatization to estrone. The granulosa cell layers have continued to differentiate from each other. The membrana layer, through the action of FSH, acquires LH receptors. This differs from the cumulus layer, which lacks LH receptors. The final progression to a mature graafian follicle is a selection process that in most cases generates one dominant follicle destined for ovulation.
The selection process actually begins in the midluteal phase of the previous cycle. The rise in estrogen level that is generated by the preovulatory follicle augments FSH activity within the follicle while exerting negative feedback on the pituitary release of FSH. The decrease in pituitary release of FSH results in withdrawal of gonadotropin support from the smaller antral follicles, promoting their atresia. The dominant follicle continues to grow, despite decreasing levels of FSH, by accumulating a greater mass of granulosa cells with more FSH receptors. Increased vascularity of the theca cells allows preferential FSH delivery to the dominant follicle despite waning FSH levels. Increased estrogen levels in the follicle facilitate FSH induction of LH receptors on the granulosa cells, allowing the follicle to respond to the ovulatory surge of LH levels. Without estrogen, LH receptors do not develop on the granulosa cells.
The generation of the LH surge is absolutely required for ovulation and oocyte maturation. The amplified production of LH in the midcycle is due to an increase in pituitary sensitivity to GnRH. The sensitization is mediated by the positive feedback effect of the exponential rise in estrogen and possibly inhibin A. This surge results in the resumption of meiosis I in the oocyte with release of a polar body just prior to ovulation. Evidence suggests that the granulosa membrana cells secrete an oocyte maturation inhibitor (OMI), which interacts with the cumulus to block the progression of meiosis during most of folliculogenesis. It is theorized that OMI exerts its inhibitory influence by stimulating the cumulus to increase cAMP production, which diffuses into the oocyte and halts meiotic maturation. The LH surge overcomes the arrest of meiosis by inhibiting OMI secretion; thereby decreasing cAMP levels and increasing intracellular calcium, allowing the resumption of meiosis (Figure 13–8).
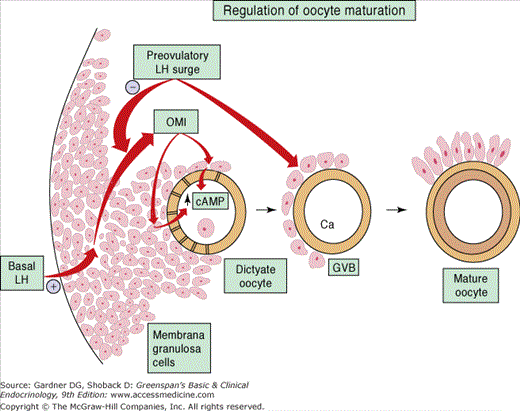
Just prior to the ovulation, progesterone production increases, and this may be responsible, at least in part, for the midcycle peak in FSH and the coordination of the LH surge. The FSH peak stimulates the production of an adequate number of LH receptors on the granulosa cells for luteinization. FSH, LH, and progesterone induce expression of proteolytic enzymes that degrade the collagen in the follicular wall, thereby making it prone to rupture. Prostaglandin (PG) production increases and may be responsible for contraction of smooth muscle cells in the ovary, aiding the extrusion of the oocyte.
The LH surge lasts approximately 48 to 50 hours. Thirty-six hours after onset of the LH surge, ovulation occurs. The feedback signal to terminate the LH surge is not known. Perhaps the rise in progesterone production results in a negative feedback loop and inhibits pituitary LH secretion by decreasing the pulsatility of GnRH. In addition, just prior to ovulation LH downregulates its own receptors, which decreases the activity of the functional hormonal unit. As a result, estradiol production decreases.
Following ovulation and in response to LH, the granulosa cells (membrana) and thecal interstitial cells that remain in the ovulated follicle differentiate into granulosa lutein and theca lutein cells, respectively, to form the corpus luteum. In addition, LH induces the granulosa lutein cells to produce vascular endothelial growth factor (VEGF), which plays an important role in developing the corpus luteum vascularization. This neovascularization penetrates the basement membrane and provides the granulosa lutein cells with LDL for progesterone biosynthesis. After ovulation, the luteal cells upregulate their LH receptors by an unknown mechanism. This is critical in that it allows basal levels of LH to maintain the corpus luteum. Rescue of the corpus luteum with hCG from the developing conceptus works through the LH receptor, which is vital for embryonic life.
Hormone production by the corpus luteum requires the cooperation of theca lutein and granulosa lutein cells, much like the preovulatory follicle (see Figure 13–7). In response to LH and hCG, the theca lutein cells increase the expression of all the enzymes responsible for androstenedione production (see earlier). Aromatase activity is increased in the granulosa lutein cells by LH to aromatize the androgens to estrogen. A notable difference in the granulosa lutein cells, as opposed to the preovulatory granulosa cells, is that LH also induces the expression of P450scc and 3β-HSD, which enables the cell to synthesize progesterone. The secretion of progesterone and estradiol is episodic and correlates with the LH pulses. FSH has minimal influence on progesterone production but continues to stimulate estrogen production during the luteal phase. The progesterone levels rise and reach a peak on approximately day 8 of the luteal phase. The luteal phase lasts approximately 14 days.
The corpus luteum starts to undergo luteolysis (programmed cell death) approximately 9 days after ovulation. The mechanism of luteal regression is not completely known. Once luteolysis begins, there is a rapid decline in progesterone levels. A number of studies suggest that estrogen has a role in luteolysis. It has been shown that direct injection of estrogen into the ovary containing a corpus luteum induces luteolysis and a fall in progesterone levels. Experimental data suggest that there is increased aromatase activity in the corpus luteum just prior to luteolysis. The rise in aromatase activity is secondary to gonadotropin (FSH and LH) stimulation, although later in the luteal phase, FSH probably plays a more important role. Consequently, estrogen production increases, and this decreases 3β-HSD activity. This may result in a decline in progesterone levels and lead to luteolysis. Furthermore, local modifiers such as oxytocin, which is secreted by luteal cells, have been shown to modulate progesterone synthesis. Other evidence supports the role of PGs in luteolysis. Experimental data suggest that PGF2α, which is secreted from the uterus or ovary during the luteal phase, stimulates the synthesis of cytokines such as tumor necrosis factor; this causes apoptosis and, therefore, may be linked to corpus luteum degeneration.
The process of luteolysis is known to involve proteolytic enzymes. Evidence suggests that matrix metalloproteinase (MMP) activity is increased during luteolysis. hCG is a known modulator of MMP activity. This may play an important role in early pregnancy, when hCG rescues the corpus luteum and prevents luteal regression. However, in the absence of pregnancy, the corpus luteum regresses, resulting in a decrease in progesterone, estradiol, and inhibin A levels. The decrease in these hormones allows for increased GnRH pulsatility and FSH secretion. The rise in FSH will rescue another cohort of follicles and initiate the next menstrual cycle.
The sole function of the uterus is to accommodate and support a fetus. Furthermore, it is the endometrium, the lining of the uterine cavity, that differentiates during the menstrual cycle so that it can support and nourish the conceptus. Histologically, the endometrium is made up of an epithelium composed of glands and a stroma that contains stromal fibroblasts and extracellular matrix. The endometrium is divided into two layers based on morphology: the basalis layer and the functionalis layer. The basalis layer lies adjacent to the myometrium and contains glands and supporting vasculature. It provides the components necessary to develop the functionalis layer. The functionalis is the dynamic layer that is regenerated every cycle. More specifically, it is this layer that can accommodate implantation of the blastocyst.
During the menstrual cycle, the endometrium responds to hormones secreted from the ovaries. Somewhat like the other endocrine organs, it contains a network of local factors that modulate hormonal activity. The endometrial phases are coordinated with ovulatory phases. During the follicular phase, the endometrium goes through the proliferative phase. It begins with the onset of menses and ends at ovulation. During the luteal phase, the endometrium undergoes the secretory phase. It starts at ovulation and ends just before menses. If implantation does not occur, a degenerative phase follows the secretory phase within the endometrium. It is this phase that results in menstruation. The next section will discuss the phases of the endometrium in more detail.
During the follicular phase, the ovary secretes estrogen, which stimulates the glands in the basalis to initiate formation of the functionalis layer. Estrogen promotes growth by enhancing gene expression of cytokines and a variety of growth factors, including EGF, TGFα, and IGF. These factors provide a microenvironment within the endometrium to modulate the effects of hormones. At the beginning of the menstrual cycle, the endometrium is thin—usually less than 2 mm in total thickness. The endometrial glands are straight and narrow and extend from the basalis toward the surface of the endometrial cavity. As the epithelium and the underlying stroma develop, they acquire estrogen and progesterone receptors. The spiral blood vessels from the basalis layer extend through the stroma to maintain blood supply to the epithelium. Ultimately, the lining (functionalis) surrounds the entire uterine cavity and achieves a thickness of 3 to 5 mm in height (total thickness 6 to 10 mm). This phase is known as the proliferative phase.
After ovulation, the ovary secretes progesterone, which inhibits further endometrial proliferation. This mechanism may be mediated by antagonizing estrogen effects. Progesterone downregulates estrogen receptors in the epithelium and mediates estradiol metabolism within the endometrium by stimulating 17β-HSD activity and converting estradiol into the weaker estrogen, estrone. During the luteal phase, the glandular epithelium accumulates glycogen and begins to secrete glycopeptides and proteins—along with a transudate from plasma—into the endometrial cavity. It is this fluid that provides nourishment to the free-floating blastocyst. Progesterone also stimulates differentiation of the endometrium and causes characteristic histologic changes. The glands become progressively more tortuous, and the spiral vessels coil and acquire a corkscrew appearance. The underlying stroma becomes very edematous as a result of increased capillary permeability, and the cells begin to appear large and polyhedral, with each cell developing an independent basement membrane. This process is termed predecidualization. These cells are very active and respond to hormonal signals. They produce PGs along with other factors that play an important role in menstruation, implantation, and pregnancy. This phase is known as the secretory phase.
If there is no implantation of an embryo, the endometrium enters the degenerative phase. Estrogen and progesterone withdrawal promotes prostaglandin production—PGF2α and PGE2. These PGs stimulate progressive vasoconstriction and relaxation of the spiral vessels. These vasomotor reactions lead to endometrial ischemia and reperfusion injury. Eventually, there is hemorrhage within the endometrium with subsequent hematoma formation. The progesterone withdrawal triggers MMP activity, which facilitates degradation of the extracellular matrix. As ischemia and degradation progress, the functionalis becomes necrotic and sloughs away as menstruum consisting of endometrial tissue and blood. The amount of blood lost in normal menses ranges from 25 mL to 60 mL. Although PGF2α is a potent stimulus for myometrial contractility and limits postpartum bleeding, it has minimal impact on cessation of menstrual bleeding. The major mechanisms responsible for limiting blood loss involve the formation of thrombin-platelet plugs and estrogen-induced healing of the basalis layer by reepithelialization of the endometrium, which begins in the early follicular phase of the next menstrual cycle.
If conception takes place, implantation can occur in the endometrium during the midsecretory (midluteal) phase, at which time it is of sufficient thickness and full of sustenance. The syncytiotrophoblast subsequently secretes hCG, which rescues the corpus luteum and maintains progesterone secretion, essential for complete endometrial decidual development.
In summary, the ovary has two phases during the menstrual cycle: the follicular phase and the luteal phase. The endometrium has three phases and is synchronized by the ovary. The complex feedback loops between the ovary and the hypothalamic-pituitary axis regulate the menstrual cycle. During the follicular phase, the ovary secretes estradiol, which stimulates the endometrium to undergo the proliferative phase. After ovulation (luteal phase), the ovary secretes estrogen and progesterone, which maintains the endometrial lining and promotes the secretory phase. In a nonpregnant cycle, luteolysis occurs, resulting in cessation of hormone production. This hormone withdrawal results in the degenerative phase and the onset of menses.
Menstrual Disturbances
The mean age of menarche is 12.8 years. Normally, this event marks the completion of puberty. The onset of regular cyclicity in the menstrual cycle is determined by the duration of the maturation process of the HPO axis, which is quite variable. As a result, anovulation can occur in 50% to 80% of girls 2 years after menarche, and in more than 20% of girls, it can persist until 5 years after menarche. This period of time can be frustrating for many girls and their parents. The evaluation should include a clinical examination and reassurance. If positive findings are noted, limited, pertinent diagnostic tests (see later) are indicated.
Amenorrhea can be defined as either the absence of menarche by age 16 or no menses for more than three cycles in an individual who has previously had cyclic menses. The definition, although arbitrary, nonetheless gives a general guideline to the clinician for further evaluation. Although amenorrhea does not cause harm, in the absence of pregnancy, it may be a sign of genetic, endocrine, and/or anatomic abnormalities. If the outflow tract is intact, amenorrhea is most likely the result of disruption in the HPO axis. These aberrations can affect any level of control in the menstrual cycle and, thus, result in menstrual abnormalities.
Amenorrhea was formerly classified as primary or secondary depending on whether the individual had experienced menses in the past. This classification may lead to misdiagnosis of the cause of amenorrhea. Although primary amenorrhea is more often associated with genetic and anatomic abnormalities, each individual should be assessed by means of the history and clinical findings, including the presence or absence of secondary sexual characteristics (Table 13–3). The causes of amenorrhea are grouped according to the level of involvement in the regulatory systems that govern normal menstrual activity (ie, hypothalamic, pituitary, ovarian, and uterine). An algorithm for the workup of amenorrhea or oligomenorrhea in the presence of secondary sexual characteristics is illustrated in Figure 13–9.
| I. Absent breast development; uterus present |
| A. Gonadal failure |
| 1. Gonadal agenesis |
| 2. Gonadal dysgenesis |
| a. 45,X (Turner syndrome) |
| b. 46,X abnormal X (eg, short- or long-arm deletion) |
| c. Mosaicism (eg, X/XX, X/XX/XXX) |
| d. 46,XX or 46,XY (Swyer syndrome) gonadal dysgenesis |
| B. Defects in estrogen biosynthesis (46, XX) |
| 1. 17,20-Lyase deficiency |
| 2. CYP17α deficiency |
| C. Hypothalamic failure secondary to inadequate GnRH release |
| 1. Insufficient GnRH secretion |
| a. FHA |
| b. Anorexia nervosa and bulimia |
| c. CNS neoplasm (craniopharyngioma, gliomas) |
| d. Excessive exercise |
| e. Constitutional delay |
| 2. Inadequate GnRH synthesis (Kallmann syndrome) |
| 3. Developmental anatomic abnormalities in central nervous system |
| D. Pituitary failure |
| 1. Isolated gonadotropin insufficiency |
| 2. GnRH resistance |
| 3. Pituitary tumors (hyperprolactinemia) |
| 4. Pituitary insufficiency |
| a. Infections (mumps, encephalitis) |
| b. Newborn kernicterus |
| 5. Prepubertal hypothyroidism |
| II. Breast development; uterus absent |
| A. Androgen resistance (androgen insensitivity syndrome) |
| B. Congenital absence of uterus (utero-vaginal agenesis) |
| III. Absent breast development; uterus absent |
| A. Defects in testosterone biosynthesis (46,XY) |
| 1. 17,20-Lyase deficiency |
| 2. CYP17α deficiency |
| 3. 17β-Hydroxysteroid dehydrogenase deficiency |
| B. Testicular regression syndrome (46,XY) |
| IV. Breast development; uterus present |
| A. Pregnancy |
| B. Hypothalamic etiology |
| 1. FHA |
| 2. Anorexia nervosa and bulimia |
| 3. Psychogenic (depression) |
| 4. CNS neoplasm |
| 5. Chronic disease |
| C. Pituitary etiology |
| 1. Pituitary tumors (hyperprolactinemia) |
| 2. Pituitary insufficiency |
| a. Hypotensive event (Sheehan syndrome) |
| b. Infections |
| c. Autoimmune destruction |
| d. Iatrogenic (surgery, radiation) |
| D. Ovarian etiology |
| 1. POF |
| a. Mosaicism (46,XX/XO,XX/XY) |
| b. Autoimmune destruction |
| c. Iatrogenic (radiation, chemotherapy) |
| d. Fragile X syndrome |
| e. Infections |
| 2. Resistant ovarian syndrome (Savage syndrome) |
| E. Chronic estrogenized anovulation |
| 1. Hyperandrogenic |
| a. PCOS |
| b. Non classical congenital adrenal hyperplasia |
| c. Cushing syndrome |
| d. Androgen secreting tumors |
| 2. Other |
| a. Adrenal insufficiency |
| b. Thyroid disorders |
| F. Outflow tract |
| 1. Congenital abnormalities |
| a. Transvaginal septum |
| b. Imperforate hymen |
| 2. Asherman syndrome |
Figure 13–9
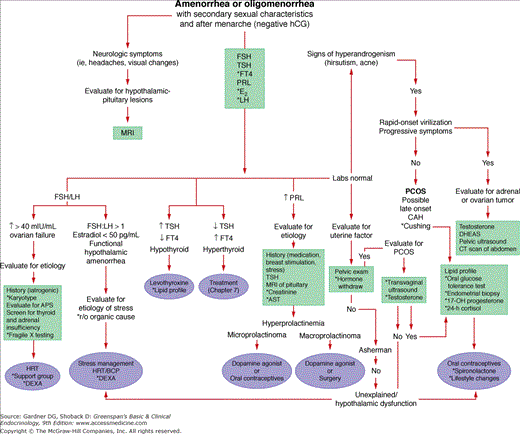
An algorithm for women who have developed secondary sexual characteristics, have experienced menarche, and are presenting with amenorrhea or oligomenorrhea. A negative human chorionic gonadotropin test documents the nonpregnant state. All women should be screened for known causes of oligomenorrhea, whether or not hyperandrogenism is present. Hyperandrogenism suggests either polycystic ovarian syndrome (PCOS) or late-onset congenital adrenal hyperplasia (CAH). If rapid in onset, progressive virilization requires an evaluation for an androgen-secreting tumor. Management of late-onset CAH and PCOS is similar. Clinical history and physical findings suggesting hypoestrogenism, stress, or thyroid dysfunction are indications for measurement of estradiol (E2), luteinizing hormone (LH), and free thyroid hormone (FT4) levels in plasma (APS, autoimmune polyglandular syndrome). *As indicated (refer to text).
The hypothalamus is the source of GnRH, which directs the synthesis and secretion of pituitary gonadotropins. Dysfunction at this level leads to hypogonadotropic hypogonadism or eugonadotropic hypogonadism. Disorders of GnRH production can result in a wide range of clinical manifestations. The individual’s appearance is dependent on the age at onset and the degree of dysfunction.
Isolated GnRH deficiency results in hypogonadotropic hypogonadism. Female patients present with amenorrhea, and females and males present with absent or incomplete pubertal development secondary to absent or diminished sex steroids (estradiol in females, testosterone in males). They have normal stature with a eunuchoidal body habitus. Because the adrenal glands are unaffected by the absence of GnRH, body hair distribution is not affected.
Several genetic lesions associated with GnRH deficiency have been described. The best characterized form of GnRH deficiency is Kallmann syndrome, which involves the Kal-1 gene as well as a number of other genes. The Kal-1 gene normally codes for anosmin, an adhesion molecule that appears to be involved in the migration of GnRH and olfactory neurons from the olfactory placode to the hypothalamus. The Kal-1 gene is located on the short arm of the X chromosome. Most cases of Kallmann syndrome are sporadic, although the disorder has also been observed to have a familial pattern, and most often it is transmitted by X-linked recessive inheritance. Autosomal recessive and dominant patterns have been reported but are much less common. When mutations exist in the Kal-1 gene, there may be associated defects, including anosmia and, less frequently, midline facial defects, renal anomalies, and neurologic deficiency. The disorder affects both sexes, but because of the X-linked inheritance pattern it is more common in boys. Unlike males, the specific genetic mutations in the Kal-1 gene in females with hypogonadotropic hypogonadism have not been identified, suggesting there are other genetic mutations that cause this disorder. Recently, it was determined that heterozygous mutations in fibroblast growth factor receptor 1 (FGFR1) gene, known as the Kal-2, were identified in cases of Kallmann syndrome. FGFR1 and Kal-1 are coexpressed at the same sites during development and are thought to have a functional interaction. Mutations in the Kiss-1 derived peptide receptor, GPR54, have also been identified in these patients. Several mechanisms of how GPR54 may regulate gonadotropin production via GnRH function or production have been proposed. Several studies have shown that females with presumed Kallmann syndrome demonstrate variable responses to exogenous GnRH administration, which suggests a GnRH receptor defect. In fact, mutations in the GnRH receptor have been identified in both sexes and are inherited in an autosomal recessive fashion.
Management of hypogonadotropic hypogonadism involves scheduled hormone replacement therapy (HRT) to stimulate the development of secondary sexual characteristics and increase bone mineral density. If pregnancy is desired, treatment involves the administration of pulsatile GnRH or gonadotropin treatment.
Functional hypothalamic amenorrhea—Functional hypothalamic amenorrhea is one of the most common types of amenorrhea and accounts for 15% to 35% of cases. It is an endocrine disorder, although the exact mechanism has not been definitively determined. It is characterized by a reduced GnRH drive (decrease in pulse frequency and amplitude), leading to low or low-normal serum levels of FSH and LH and resulting in anovulation. The ratio of serum FSH to LH in these patients is often equivalent to that of a prepubertal female with a relative FSH dominance.
The adipocyte hormone, leptin, has been implicated in the development of this disorder. Leptin is an important nutritional satiety factor, but it is also necessary for maturation of the reproductive system. The potential link to the reproductive system is thought to be through leptin receptors, which have been identified in the hypothalamus and gonadotropes. This is supported by the observation that leptin can stimulate GnRH pulsatility and gonadotropin secretion. Several studies suggest that women with functional hypothalamic amenorrhea have lower serum leptin levels in comparison with eumenorrheic controls. This relative deficiency may lead to dysfunctional release of GnRH and subsequent development of functional hypothalamic amenorrhea.
Abnormal, and often subtle, activation of the hypothalamic-pituitary-adrenal axis is associated with functional hypothalamic amenorrhea. The inciting event may be excessive production of corticotropin-releasing hormone (CRH), which has been shown to decrease the pulse frequency of GnRH and increase cortisol levels in vivo. In contrast, another study suggests that although acute elevations of CRH can suppress GnRH release, this suppression cannot be maintained with CRH alone. The cause of functional hypothalamic amenorrhea often remains unclear, but the associated hypercortisolemia suggests that it is preceded by psychologic stress, strenuous exercise, or poor nutrition. There is support for the concept that these factors may act synergistically to further suppress GnRH drive. In fact, patients with functional hypothalamic amenorrhea resulting from psychologic stress are usually high achievers who have dysfunctional coping mechanisms when dealing with daily stress. The severity of hypothalamic suppression is reflected by the clinical manifestations. The significant interpatient variability in the degree of psychologic or metabolic stress required to induce a menstrual disturbance explains the heterogeneity of clinical presentations, ranging from luteal phase defects to anovulation with erratic bleeding to amenorrhea.
Functional hypothalamic amenorrhea is reversible. Interestingly, the factors that have predicted the rate of recovery are body mass index and basal cortisol levels. When patients recover, ovulation is preceded by return of cortisol levels to baseline. Some experts have shown that cognitive behavioral therapy, teaching the patient how to cope with stress—and nutritional consultation—reverse this condition. Complete reversal may be less likely if the functional insult occurs during the period of peripubertal maturation of the HPO axis.
The diagnosis of functional hypothalamic amenorrhea can be made if the FSH-LH ratio is greater than 1 in the presence of hypoestrogenemia. However, a minor disturbance of hypothalamic dysfunction may present with normal laboratory findings, a clinical history that coincides with stress, and a negative evaluation for other causes of anovulation. Interestingly, most of these patients, despite hypoestrogenism, do not have symptoms. The estrogen status should still be evaluated given the strong correlation between hypoestrogenemia and the development of osteoporosis. Estrogen status can be determined by means of the progesterone withdrawal test or by measurement of serum estradiol (<50 pg/mL). If there is no withdrawal or the estradiol is less than 50 pg/mL, HRT with combination contraceptive hormones (or traditional HRT) should be instituted. If withdrawal bleeding occurs, any cyclic progestin-containing therapy is adequate to combat unopposed estrogen and the development of endometrial hyperplasia.
Amenorrhea in the female athlete—Hypothalamic dysfunction has been observed in female athletes. Competitors in events such as gymnastics, ballet, marathon running, and diving can show menstrual irregularities ranging from luteal phase defects to amenorrhea. The female athletic triad as defined by the American College of Sports Medicine is characterized by disordered eating, amenorrhea, and osteoporosis. The associated nutritional deficiencies can lead to impaired growth and delayed sexual maturation. The neuroendocrine abnormalities are similar to those of women with functional hypothalamic amenorrhea.
These patients have very low body fat, often below the tenth percentile. There is evidence that a negative correlation between body fat and menstrual irregularities exists. In addition, there appears to be a critical body fat level that must be present in order to have a functioning reproductive system. Several studies have shown that these amenorrheic athletes have significantly lower serum leptin levels, which further supports leptin’s role as a mediator between nutritional status and the reproductive system. The strenuous exercise these athletes engage in amplifies the effects of the associated nutritional deficiency. This synergism causes severe suppression of GnRH, leading to the low estradiol levels.
Amenorrhea alone is not harmful. However, low serum estradiol over a period of time may lead to osteoporosis and delayed puberty. An analysis of estrogen status may be obtained with measurement of serum estradiol levels or with the progestin withdrawal test (see earlier). If estrogen is low, bone mineral density (BMD) should be assessed by dual-energy x-ray absorptiometry (DXA) scan. All patients diagnosed with female athletic triad need combination contraceptive therapy or HRT.
Amenorrhea associated with eating disorders—Anorexia nervosa is a disorder characterized by relentless dieting in pursuit of a thin body habitus. Approximately 95% of cases occur in females, and the onset is chiefly in adolescence. The clinical features include extreme weight loss leading to a body weight less than 85% of normal for age and height, a distorted body image, and intense fear of gaining weight. These patients usually have a preoccupation with food and are hyperactive, with an obsessive-compulsive personality. The associated symptoms include hypothermia, mild bradycardia, dry skin, constipation, and symptoms of hypoestrogenemia. Furthermore, as part of the diagnostic criteria, they must experience at least 3 months of no menses.
The dysfunction in the neuroendocrine system is similar to but often more severe than that described in association with functional hypothalamic amenorrhea. The severe reduction in GnRH pulsatility leads to suppression of FSH and LH secretion, possibly to undetectable levels, and results in anovulation and low serum estradiol levels. Given the severe psychologic and metabolic stress experienced by these individuals, the hypothalamic-pituitary-adrenal axis is activated. The circadian rhythm of adrenal secretion is maintained, but both cortisol production and plasma cortisol levels are persistently elevated secondary to increased pituitary secretion of adrenocorticotropic hormone (ACTH). Serum leptin levels in these individuals are significantly lower than normal healthy controls and correlate with percentage of body fat and body weight. A rise in leptin levels in response to dietary treatment is associated with a subsequent rise in gonadotropin levels. This further suggests leptin’s role as a potential link between energy stores and the reproductive system.
The self-induced starvation state associated with anorexia nervosa leads to additional endocrine abnormalities not observed in other causes of hypothalamic amenorrhea. For instance, thyroid hormone metabolism is altered. TSH and T4 levels are in the low-normal range, but T3 levels are usually below normal. This is attributable to decreased peripheral conversion of T4 to T3 and increased conversion of T4 to the metabolically inactive thyroid hormone, reverse T3—a change that often resembles other states of starvation. This may be a protective mechanism in that the relative hypothyroid state attempts to reduce basal metabolic function in response to a highly catabolic state.
Bulimia occurs in about half of patients with anorexia and is defined as binge eating followed by self-induced purging. Not all bulimics have low body weight—in fact, normal-weight bulimic individuals are much more common. These patients also have a variety of neuroendocrine aberrations—often to a lesser degree than those with anorexia—which also lead to menstrual disturbances. Leptin levels are lower than in matched controls but not as low as in individuals with anorexia nervosa. They also have neurotransmitter abnormalities—notably low serotonin levels—which might help explain the often coexisting psychologic difficulties.
Anorexia nervosa is a life-threatening illness with a significant mortality rate due to its metabolic consequences. Anorexic patients should be considered for inpatient therapy and management with a multidisciplinary approach that includes nutritional counseling and psychotherapy. Force-feeding may be necessary in some patients. If weight gain cannot be achieved with oral intake, meals may need to be supplemented by enteral or parenteral feeding. Because anorexia nervosa is a hypoestrogenic state and there is a high potential for the development of osteoporosis, all patients should receive hormone therapy either in the form of HRT or combination contraceptive pills. It is important to note that many patients with anorexia will continue to have reduced bone size regardless of sex steroid administration presumably due to the concurrent malnutrition and metabolic compromise. Weight rehabilitation and recovery is associated with bone accretion; however, the loss of bone may not be completely reversible.
In summary, the hypothalamic amenorrhea endocrine syndromes are probably a continuum of disordered eating and nutritional deficiencies resulting in increasingly severe abnormalities in the reproductive system. Furthermore, the age at onset affects the potential complications of these disorders. If low estradiol levels are present before age 20, bone mineralization may be profoundly affected, because this period is critical for building peak bone mass. In addition, if these conditions occur prior to puberty, it may result in stunted growth and delayed development of secondary sexual characteristics.
Numerous anatomic abnormalities within the central nervous system can result in menstrual disturbances. These include developmental defects, brain tumors, and infiltrative disorders. The most common anatomic lesion associated with delayed puberty and amenorrhea is a craniopharyngioma. It is derived from Rathke pouch and extends into the hypothalamus, pituitary, and third ventricle. The symptoms include headaches, visual loss, and hypoestrogenism.
Infiltrative disorders that involve the hypothalamus are uncommon but can result from systemic diseases, including sarcoidosis, histiocytosis, hemochromatosis, and lymphoma. These diseases do not initially present with amenorrhea. However, in the presence of these diseases, the hypothalamus may be affected, so they should be part of the differential diagnosis of amenorrhea.
There are a few genetic mutations affecting the pituitary that cause amenorrhea. Rare autosomal recessive mutations may cause deficiencies in FSH, LH, TSH, prolactin (PRL), and growth hormone (GH). The clinical manifestations may include delayed puberty, a hypoestrogenic state, and infertility.
A deficiency in FSH and LH may be a result of GnRH receptor gene mutations. Such mutations are primarily compound heterozygous mutations that affect GnRH receptor-dependent signal transduction. The phenotype of these individuals is similar to that of those with isolated GnRH deficiency. In fact, some investigators speculate that these receptor mutations may be the cause of isolated GnRH deficiency in women, given that no mutations have yet been identified in the ligand or Kal-1 gene. The estimated prevalence of GnRH receptor mutations in women with hypothalamic amenorrhea is 2%. In a family with other affected females, the prevalence is 7%.
Other rare genetic defects have been associated with amenorrheic women. Mutations in the FSHβ gene have been reported. These have an autosomal recessive pattern of inheritance and lead to low serum FSH and estradiol levels and high plasma LH levels. The clinical features include minimal development of secondary sexual characteristics and amenorrhea with no history of menses. Combined hormone deficiencies have also been described. Mutations in Prop-1, a pituitary transcription factor for Pit-1, lead to deficiencies in gonadotropins, TSH, PRL, and GH. These patients present with stunted growth, hypothyroidism, and delayed puberty in addition to amenorrhea.
Hyperprolactinemia is one of the most common causes of amenorrhea, accounting for 15% to 30% of cases. In the absence of pregnancy or postpartum lactation, persistently elevated PRL is almost always associated with a hypothalamic-pituitary disorder. Normal PRL secretion is regulated by several stimulatory and inhibitory factors (see Chapter 4). PRL secretion is primarily under tonic inhibition by dopamine, so that any interference with dopamine synthesis or transport from the hypothalamus may result in elevated PRL levels. In addition to menstrual disturbances, individuals with hyperprolactinemia may present with galactorrhea. In fact, hyperprolactinemia is a common cause of galactorrhea, and up to 80% of patients with amenorrhea and galactorrhea have elevated PRL levels. Other associated symptoms include headaches, visual field defects, infertility, and osteopenia.
The mechanism whereby hyperprolactinemia causes amenorrhea is not completely known. Studies have shown that PRL can affect the reproductive system in several ways. PRL receptors have been identified on GnRH neurons and may directly suppress GnRH secretion. Others have postulated that elevated PRL levels inhibit GnRH pulsatility indirectly by increasing other neuromodulators such as endogenous opioids. There is also evidence that GnRH receptors on the pituitary may be downregulated in the presence of hyperprolactinemia. Furthermore, PRL may affect the ovaries by altering ovarian progesterone secretion and estrogen synthesis. The best data now available suggest that hyperprolactinemia causes amenorrhea primarily by suppression of GnRH secretion.
Approximately half of patients with elevated PRL levels have radiologic evidence of a pituitary tumor. The most common type is a PRL-secreting tumor (prolactinoma), accounting for 40% to 50% of pituitary tumors. Prolactinomas are composed mainly of lactotrophs, and they secrete PRL. Occasionally these tumors secrete both GH and PRL.
The diagnosis of a pituitary adenoma is usually made by examination of the pituitary with magnetic resonance imaging (MRI). These tumors are categorized into two groups based on their dimensions—microadenomas are those less than 10 mm in diameter and macroadenomas are larger than 10 mm. These tumors are usually located in the lateral wings of the anterior pituitary. Microadenomas are typically wholly contained within the confines of the pituitary gland. Very rarely, a microadenoma infiltrates the surrounding tissue, including the dura, cavernous sinus, or adjacent skull base. A macroadenoma may expand farther and grow out of the sella to impinge on surrounding structures, such as the optic chiasm or may extend into the sphenoid sinus. As a result, macroadenomas are more frequently associated with severe headaches, visual field defects, and ophthalmoplegia. The incidence of a microadenoma progressing to a macroadenoma is relatively low, only 3% to 7%. During pregnancy the risk of a microprolactinoma enlarging is also low, but in the presence of a macroprolactinoma, the chance of tumor growth is up to 25%.
Some investigators have found a correlation between pituitary adenoma size and serum PRL levels. If the serum PRL is less than 100 ng/mL, a microprolactinoma is more likely, whereas if it is greater than 100 ng/mL, a macroprolactinoma is often present. Although this correlation has been reported, the evidence supporting it is not strong. In fact, low PRL levels may also be associated with nonfunctioning macroadenomas. This is thought to reflect impingement of the adenoma on the pituitary stalk leading to reduced dopamine secretion and loss of the tonic suppression of prolactin secretion. These nonfunctioning tumors may synthesize glycoproteins such as FSH, LH, or their free alpha subunits. Rarely, functioning tumors may arise from other pituitary cells, resulting in excessive hormone secretion. If a macroadenoma is present, measurement of IGF-I, alpha subunit, TSH, and 24-hour urinary cortisol will begin to exclude other functioning adenomas.
Other tumors of nonpituitary origin may also result in delayed puberty and amenorrhea. The most common of these is a craniopharyngioma. Although craniopharyngiomas are commonly located in the suprasellar region, they can also involve the sella. These tumors have not been shown to produce hormones, but because they may compress the infundibulum, they can interfere with the tonic inhibition of PRL secretion and result in mildly elevated PRL levels.
Hyperprolactinemia in a patient with amenorrhea is defined as a PRL level greater than 20 ng/mL, although the limit of normal may vary between laboratories. Normal PRL release follows a sleep-circadian rhythm, but PRL may also be secreted in response to stress, physical exercise, breast stimulation, or a meal. Therefore, PRL should be measured in the midmorning hours and in the fasting state. Other causes of mildly elevated PRL include medications such as oral contraceptives, neuroleptics, tricyclic antidepressants, metoclopramide, methyldopa, and verapamil. Hyperprolactinemia has also been observed in several chronic diseases, including cirrhosis and renal disease. Furthermore, inflammatory diseases such as sarcoidosis and histiocytosis can infiltrate the hypothalamus or pituitary and result in hyperprolactinemia. Elevated PRL levels may be a physiologic response. During pregnancy, PRL levels may be two to four times baseline. With postpartum breast-feeding, the PRL level should be below 100 ng/mL after 7 days and below 50 ng/mL after 3 months. If a woman is not breast-feeding, PRL levels should return to baseline by 7 days postpartum.
Persistently elevated PRL levels may also be present in primary hypothyroidism. Approximately 40% of patients with primary hypothyroidism present with a minimal increase in prolactin (25-30 ng/mL), and 10% present with even higher serum levels. Individuals with primary hypothyroidism have an increase in thyrotropin-releasing hormone (TRH) from the hypothalamus, which stimulates TSH and PRL release and leads to hyperprolactinemia. Patients with long-standing primary hypothyroidism may eventually manifest profound pituitary enlargement due to hypertrophy of thyrotrophs. This mass effect with elevated PRL levels mimics a prolactinoma. Therefore, all patients with hyperprolactinemia should have their thyroid function investigated to exclude hypothyroidism as the cause.
Prolactinomas are the most common cause of persistent hyperprolactinemia. All patients with elevated PRL levels should have the test repeated for verification. In addition to blood tests, a careful clinical and pharmacologic history and physical examination should be performed to exclude other causes of hyperprolactinemia. If elevated PRL levels persist or if any measurement is found to be above 100 ng/mL, MRI of the hypothalamic-pituitary region should be performed. If a microadenoma is observed, the diagnosis of microprolactinoma can be made. If a macroadenoma is observed, other pituitary hormones should be measured to exclude other functioning adenomas or hypopituitarism. All patients diagnosed with macroadenoma should have a visual field examination.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


