Chapter 29
Feeding Children with Neurodisabilities
Jennifer Douglas and Leanie Huxham
Introduction
Neurodisability is a term used to describe conditions affecting the brain and central nervous system (CNS) and includes muscular, developmental, motor, sensory, learning and neuropsychiatric disorders. CNS damage can be due to disease, genetics, oxygen deprivation or acquired brain injury, amongst other causes, and can occur at any stage in a child’s life. The majority of research is on children with motor disorders and cerebral palsy, which are the two most common causes of neurodisability [1]. These children often have neurological involvement of other body systems as part of their condition [2]. In the past 10 years there has been a steady increase in the number of children with severe disability due to increased survival of preterm infants and better survival outcomes for children with brain injury. The EPICure study has shown that between 1995 and 2006 survival rate has improved for preterm infants born at or before 26 weeks. Survival rate in the UK for preterm infants born at 26 weeks is 78%, but many of these have neurodevelopmental disability [3, 4].
Many children with neurodisability have difficulties with eating and drinking, and they are likely to have nutritional concerns that need to be addressed [1]. Oromotor dysfunction is associated with poor health and nutritional outcomes, affecting up to 90% of children with moderate to severe cerebral palsy [5]. Those with motor, physical or sensory impairments are more likely to struggle [2] and the more severe the disability, the more likely the child is to be at nutritional risk [1, 6]. The ability of infants, children and adolescents to achieve their potential for growth and development depends on the intervention provided at critical time periods. Many children with neurological impairment would benefit from individual nutritional assessment and management as part of their overall care [7]. It has been accepted in the past that children with neurodisability, especially those with CP, are small as part of their condition. With the evolution of enteral feeding it has become evident that children with neurodisability have the potential to grow if adequate nutrition is provided.
Cerebral palsy
Cerebral palsy (CP) is defined as ‘a persistent (but not unchanging) disorder of movement and posture, as the result of one or more non progressive abnormalities in the brain, before its growth and development are complete. Other clinical signs may be present as well’ [[8]]. The children more severely affected are likely to have multiple comorbidities including sensory impairments (vision, hearing, touch), perceptual difficulties (resulting in impaired sensory interpretation), learning disabilities, limited communication and medical conditions such as respiratory difficulties, seizure disorders and gastro-oesophageal reflux (GOR). There is currently no test before birth to identify CP. The incidence in the UK is currently 1 in 400 children and 2.1 per 1000 live births [9].
CP is a condition where there may be abnormal brain development or brain injury during development. This can occur before, during or after birth, and even during early childhood. It is not unusual for children to be diagnosed at a later stage when the child’s motor development is almost complete. Even before diagnosis is established children may already be experiencing oromotor problems.
Causes of CP may be complex with no obvious single cause. However, there are certain risk factors that may contribute: infection during early pregnancy; oxygen deprivation to the brain due to birth complications or cerebrovascular event in childhood; restricted intrauterine growth; blood disorder (very rare platelet abnormalities); twins or multiple birth; mother’s age over 40; low birthweight (<1.5 kg); premature birth (<37 weeks); fertility treatments, severe jaundice and kernicterus, infections to the brain and brain injury during childhood [10–12].
There are four main types of CP, which correspond to injuries to different parts of the brain [13]. Children with:
- spastic CP have permanent increased muscle tone and impairment of voluntary movement or posture due to non progressive damage to the immature brain
- hypotonic CP have low muscle tone with little or no resistance to movement. These children frequently have feeding difficulties
- athetoid CP have some loss of control of their posture and they tend to make unwanted movements. They have a mixture of high and low muscle tone and often have a high requirement for energy
- ataxic CP usually have problems with balance. They may also have shaky hand movements and irregular speech. Their muscle tone tends to be low, but can fluctuate
A child with CP may have one distinct type or, more commonly, a mixture of these. The distribution of CP can be limited to one limb (monoplegia), diplegia with two limb involvement or quadriplegia with all four limbs affected.
Factors to consider when assessing patients with CP:
- type of CP and limb involvement
- mobility: include information about locomotion such as use of wheelchair or a walker
- gastrointestinal problems: GOR and chronic constipation are common in children with CP [14]
- dependence on feeding: ability to self-feed, dependent or partially dependent on assistance from parents/carers
- feeding dysfunction: detailed information regarding how the child manages food textures such as puréed, mashed or chopped foods [15]
- feeding skills: should be assessed at regular intervals in order to identify children at nutritional risk [16] (assessment of feeding skills in Down’s syndrome are relevant for children with CP). Parents/carers should be asked about the presence of tongue thrust, drooling/dribbling causing fluid loss, lack of hand to mouth coordination, poor lip seal causing food or fluid loss, inability to communicate hunger, prolonged feeding times and dysphagia [17–19]
- environment: environmental factors can have an impact on growth and nutrition, e.g. the child’s living situation, mealtime environment, lifestyle, involvement in therapy groups, school, respite care arrangements [20]. Information about the family and the child’s experience with oral feeding should be sought as this may affect decisions about future oral or non oral feeding methods
- other medical conditions and respiratory health: these children are vulnerable to respiratory morbidity for several reasons so it is important to find out about frequency of chest infections and other respiratory problems that may have an effect on energy requirements [21]
- bone health: there is a higher risk of low bone mineral density, osteopenia and osteoporosis due to reduced weight bearing activity or being bed bound; lack of nutrition; medication limiting vitamin D metabolism; limited sun exposure
- anthropometry: there is no universally accepted method for measuring children with CP. Alternatives to measuring height are available and are discussed later (p. 780).
Improved nutritional status in children with CP is associated with improvements in general health, such as decreased irritability and spasticity, improved healing of pressure sores and improved circulation. In contrast, undernutrition is significantly associated with poor functional status, poor motor function, reduced communication ability as well as increased dependence on the carer for feeding [18]. A large multicentre study of 230 children and young people with moderate to severe CP living in the community showed that the level of feeding dysfunction was directly related to the degree of undernutrition and even those who had mild feeding dysfunction had poor growth and limited fat stores [8]. Therefore, a child requiring any modified consistency of food and fluids can be at risk of nutritional compromise. It has also been documented that 89% of children with CP need help with feeding and 56% regularly choke during mealtimes [1]. Almost a third of children with CP were found to have a height for age <25th centile in a study by Vik et al. whilst 7% were classified as being obese (weight >99.6th centile) [15].
Low micronutrient intake and low micronutrient serum concentrations are common in children with CP [22]. Nutritional status should be assessed frequently to ensure that micronutrient requirements are met. Tube feeding and the use of nutritional supplements are associated with higher micronutrient concentrations [23]. Improving nutritional status is important to improve motor function with weight gain, especially fat free mass gain, showing the most benefit [14]. It is important to take into account the type of CP the child is diagnosed with (including other comorbidities) as this has significant implications on the chosen nutritional management plan. Children with spastic quadriplegic CP, hypotonic patients and those having seizures often have significant feeding difficulties [24].
Improved nutritional status is associated with improved feeding ability and undernourished children may have poorer feeding skills. A study of 90 children with CP showed feeding competence was positively correlated to weight, triceps skinfold and mid arm circumference [25]. Findings from a more recent study on feeding methods and health outcomes concluded that oral interventions for children with CP may promote oromotor function; however, they may not be effective in promoting feeding efficiency or weight gain. The author recommended gastrostomy feeding as an alternative for children with severe feeding and swallowing problems, as well as poor weight gain [26]. Parents/carers need to know that oral feeding competence may improve (depending on the patient’s ability) by improving nutritional status and it is important to communicate this to them when making decisions about feeding methods, especially when considering artificial methods such as gastrostomy feeding.
Down’s syndrome
The incidence of Down’s syndrome in the UK is estimated at approximately 1 per 1000 live births, accounting for 725 births in 2011 [27]. Due to prenatal screening this figure has remained relatively constant, despite the rise in maternal age, with 92% of women receiving the diagnosis prenatally opting for a termination [28]. Median life expectancy has improved for people with Down’s syndrome, rising from 25 years in 1983 to 49 years in 1997, with some people living into their 60s [16].
Down’s syndrome (also called Trisomy 21) is the most common genetic cause of learning disabilities [16]. Many children with Down’s syndrome are born with congenital abnormalities such as heart defects (44%–58%), gastrointestinal problems (4%–10%), recurrent respiratory infections, joint problems, endocrine dysfunction particularly hypothyroidism, constipation and GOR. A higher incidence of coeliac disease and haematological cancers has also been found [29]. Regular screening is recommended for children with Down’s syndrome to enable early identification of the common comorbidities.
Down’s syndrome is associated with anatomical facial abnormalities, which can have significant effects on feeding. Feeding skills should be assessed at regular intervals [16]. Dysfunction occurs due to poor neuromotor control, dental anomalies, orofacial dysmorphology and hypotonia particularly of the tongue and lips. Children with Down’s syndrome are frequently mouth breathers due to narrowing of the nasal cavity and chronic inflammation of the tonsils as a result of an increased incidence of upper respiratory infections. The palate is often short and narrow, and this underdevelopment of the maxilla may alter the position of the muscles used for chewing. Infants with Down’s syndrome may have difficulty in initiating a suck and have a weak lip seal, poor coordination of the suck/swallow/breathing sequence with early fatigue, as well as having jaw instability [30]. Breast feeding is possible for infants with Down’s syndrome, but for many it will be more difficult to establish due to the associated oromotor difficulties. Adequate support is needed to achieve successful breast feeding [31]. Down’s syndrome is associated with impaired immune function and higher incidence of obesity; therefore breast feeding may confer long term benefits. Comorbidities such as congenital heart disease and gastrointestinal dysmotility may further compromise feeding [32].
Parental surveys suggest that 60% of children are totally independent feeders by early childhood and the most common problems are slight oral hypotonia, tongue thrust, difficulties in chewing, poor lip seal, and choking and gagging on food [33]. However, it is noted that this feeding success may be partly as a direct result of feeding programmes and not simply a natural developmental step, thus reinforcing the need for assessment and management programmes. Hopman et al. noted that solids were introduced at a later stage compared with controls, possibly due to low parental expectations of developmental ability [34]. Foods requiring less chewing were given, further inhibiting oromotor development. Children with Down’s syndrome often have increased oral sensitivity, interfering with the acceptance of new foods, and a high incidence of aspiration, which is possibly related to the high incidence of respiratory disease [35].
Anthropometric assessment of children with Down’s syndrome is complicated because of the associated abnormalities related to growth such as short stature, decreased head circumference and altered growth patterns. Cronk suggests that while height in people with Down’s syndrome is significantly lower than the norm, the period in which most significant growth failure occurs is during the first 5 years of life [36, 37]. Growth rate is reduced by a fifth between the ages of 3 and 36 months in both sexes. Longitudinal studies corroborate this, but show that growth velocity of children aged 7–18 years was not significantly different to the norm [38]. There are specific charts for children with Down’s syndrome [39]; they describe typical growth, not necessarily ideal growth.
Despite an energy intake below the normal requirement there is a high prevalence of obesity in children and adults with Down’s syndrome [40–42]. A 10%–15% lower resting metabolic rate, but equivalent expenditure above resting, has been found in prepubescent children with Down’s syndrome [37]. Crino et al. report that 66% of pubertal children with Down’s syndrome were obese [43]. As obesity is prevalent in adolescence and adulthood it is recommended that growth charts are used in conjunction with body mass index (BMI), using the same cut-offs for obesity as are applied to the general paediatric population. With appropriate support around nutrition and exercise obesity can be managed successfully [44].
Some studies have shown deficiencies and/or risk of deficiency of vitamin A, vitamin C, vitamin E, zinc, calcium and other nutrients in children with Down’s syndrome [41, 45–48]. It is important to note that the majority of these studies have small sample sizes and questionable study methods. The general consensus is that the majority of children with Down’s syndrome can meet vitamin and mineral requirements, but monitoring may be needed for those who are unable to eat a healthy balanced diet [49].
Neuromuscular and progressive neurological disorders
There are approximately 60 different types of muscular dystrophy and related neuromuscular conditions. Congenital neuromuscular disorders in children include spinal muscular atrophy and muscle disorders such as Duchenne muscular dystrophy and congenital muscular dystrophies. These conditions are characterised by the loss of muscle strength as progressive muscle wasting or nerve deterioration occurs.
Progressive neurological disorders are extremely rare conditions where deterioration advances over time. There are a large number of different diagnoses, e.g. Batten’s disease, leukodystrophies, Cockayne syndrome and Rett syndrome. Some conditions progress at a steady rate and others degenerate in phases.
Due to the vast number of different neuromuscular and progressive neurological disorders it is essential to conduct a literature search at the time of patient review for the most up to date information. Table 29.1 shows some of the more common disorders referred to the dietitian and some of the nutritional considerations associated with each disorder.
Table 29.1 Neuromuscular and progressive neurological disorders and nutritional considerations
| Common conditions | Nutritional considerations | Useful websites |
| Lysosomal storage disease (Tay Sachs, Fabry, Pompe, metachromic leukodystrophy, Krabbe, Canavan, Zellweger) | Commonly have short stature, skeletal deformities, muscle weakness or lack of control (e.g. ataxia, seizures), neurological failure/decline and/or loss of gained development. Poor swallow may require tube feeding or texture modified diet. Can have both undernutrition and overnutrition. Frequent dietetic monitoring required due to progressive nature of the disease [1] | www.ninds.nih.gov www.lysosomallearning.com www.rarediseases.org www.ulf.org |
| Adrenoleukodystrophy (see Chapter 20) | Lorenzo’s oil can be used in children who are asymptomatic and this may delay onset of symptoms. Symptom control is needed for all other children [1] | www.ninds.nih.gov www.rarediseases.org www.ulf.org |
| Rett syndrome | Growth failure, oromotor dysfunction, gastro-oesophageal reflux, constipation and low bone density are common. Often need assistance with feeding. May need gastrostomy feeding to improve growth [1] | www.rettuk.org www.reverserett.org.uk www.ninds.nih.gov www.rarediseases.org |
| Duchenne muscular dystrophy | Predominantly affects males. High prevalence of obesity related to lower resting energy expenditure, steroid use and reduced physical activity. Muscle wasting, poor feeding and poor swallow coordination can lead to malnutrition as they get older. Assistance is often needed with oral feeding and texture modification may improve intakes. Tube feeding may be required. Will need supplementary calcium and vitamin D if they are on steroids. Regular dietetic monitoring required [1] | www.ninds.nih.gov www.rarediseases.org www.muscular-dystrophy.org www.nlm.nih.gov www.dfsg.org.uk |
| Spinal muscular atrophy | Muscle weakness and bulbar dysfunction lead to swallowing difficulties. Respiratory problems are common including aspiration pneumonia. Undernutrition and overnutrition can occur and regular dietetic monitoring is essential. Undernutrition needs aggressive therapy to avoid exacerbation of pre-existing weaknesses. Gastro-oesophageal reflux is common and may require anti-reflux surgery or jejunal feeding. Constipation and delayed gastric emptying can commonly occur [1] | www.jtsma.org.uk www.smafoundation.org www.actsma.co.uk www.ninds.nih.gov www.lysosomallearning.com www.rarediseases.org |
| Prader–Willi syndrome (see Chapter 30) | At birth the infant typically has low birth weight for gestation, poor muscle tone (‘floppy’), difficulty sucking and may require tube feeding to prevent faltering growth. Feeding difficulties resolve over time and at 2–5 years of age hyperphagia sets in and lasts throughout the lifetime, leading to obesity if not correctly managed [1] | www.pwsa.co.uk www.praderwillisyndrome.org.uk www.geneticdiseasefoundation.org www.fpwr.ca www.pwsausa.org www.fpwr.org |
| Cockayne syndrome | Premature ageing and short stature. Growth failure and likely to have feeding problems especially as an infant. Will need tube feeding to treat faltering growth and/or manage poor swallow coordination. Likely to have developmental delay and increased tone/spasticity. Regular dietetic monitoring needed due to progressive nature of the disease [1] | www.amyandfriends.org www.cockayne-syndrome.net www.cockaynesyndrome.net www.ncbi.nlm.nih.gov |
| Batten’s disease | A neurodegenerative disease. Feeding becomes more difficult with resulting poor weight gain and frequent symptoms of aspiration or difficulty coordinating swallowing (due to progressing lack of coordination and progressive poor muscle tone). Most children will eventually need tube feeding. If they manage to feed orally they are likely to need assistance due to abnormal limb movement and poor muscle strength. Frequent dietetic monitoring is required to support child at various stages of disease progression | www.bdfa-uk.org.uk www.ninds.nih.gov www.bdsra.org www.nathansbattle.com www.hideandseek.org www.battens.org.au |
Nutritional requirements
Energy
Disabled children often have lower resting energy expenditures due to reduced mobility so their total energy requirements are often lower than the norm. Prediction equations and dietary reference values therefore tend to overestimate needs [50–52]. This has been shown by the Oxford Feeding Study research team, where disabled children tended to lay down stores of fat rather than muscle when gastrostomy fed beyond their requirements [53, 54]. Krick et al. have proposed a complicated formula using muscle tone, activity and growth to calculate nutritional requirements [55]. There is currently no universally accepted formula for predicting energy requirements in children with neurodisability. Requirements are based on individual clinical assessment and degree of mobility.
Children with neurodisability are smaller than their non disabled counterparts and until further research becomes available, dietetic consensus opinion is to use height age (a crude estimation of bone age) as a basis to estimate nutritional requirements. This should be adjusted depending on whether weight loss or weight gain is needed. A child entering their pubertal growth spurt will require more energy than one who is not. In practice energy requirements are often no more than 75% of the estimated average requirement (EAR) for height age and are often considerably less; a child receiving as little as 20–30 kcal/kg/day (85–125 kJ/kg/day) can still gain weight. The exceptions to this are those children with mixed CP which includes an athetoid component; excessive involuntary movements make their energy requirements higher and closer to the EAR for chronological age.
Protein and micronutrients
There are no studies informing the requirement for these nutrients. However, as disabled children tend to be smaller than their non disabled peers, reference nutrient intakes (RNI) for actual age are likely to be too high. Height age may be used as a basis for estimations and it is advisable to meet at least the lower reference nutrient intakes (LRNI).
Fluid
Actual body weight is used to calculate fluid requirements and can be based on general recommendations (p. 16) . However, it is interesting to note that many children appear well hydrated on fluid intakes that are lower than those derived by calculation and 75% of estimated fluid requirements may be adequate to maintain hydration.
Fibre
Currently there are no UK recommendations for fibre intake in children. In the absence of these, dietitians may refer to American recommendations which suggest the calculation of fibre intake in grams: age (years) + 5 g–10 g [56].
Nutritional assessment
Anthropometry
There are no randomised controlled trials on dietetic assessment to ascertain nutritional status in children with neurodisability. Thus the following information has been developed as a consensus guideline of best practice based on the most current literature [57]. Most research has been conducted on children with CP; therefore, caution must be taken when extrapolating these recommendations to children with other neurodisabilities.
Weight
Weight should be measured routinely on the most appropriate weighing equipment for the individual child. These include hoist scales, wheelchair scales and sitting scales for the child, as well as the carer holding the child on the scales and then their weight being subtracted. There is no evidence comparing the accuracy of the various weighing methods; thus all should be accepted as of equal value. The chosen method should be recorded and used when subsequently weighed. Weight measures should be plotted on growth charts, the frequency depending on local practice and the child’s individual circumstances, but should be a minimum of every 6 months for older children and young people but more frequently for children under the age of 2 years.
Height
Accurate height measures are often difficult to obtain in disabled children and young people, due to scoliosis or kyphosis caused by a twisted posture or contractures of the spine. A standing height is preferable, but where this is not possible a supine length is an acceptable second choice. It is important to note, however, that a supine length will measure longer than a standing height and serial measures should not be confused.
Where a length or height is not possible there are three suggested alternatives; upper arm length, lower leg length and knee height all of which have been found to correlate with actual height [58]. The frequency of measurements will depend on local practice and individual circumstances; height should be taken a minimum of every 6 months and plotted on a growth chart.
Upper arm length
Upper arm length (UAL) is measured from the acromion to the head of the radius (Fig. 29.1). It should be measured on the right or the least affected side. Two measurements are taken and then averaged. Research suggests it can only be taken accurately using an anthropometer. The measurement can be converted into a height measure and plotted on a growth chart, using the following formula [58]:
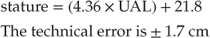
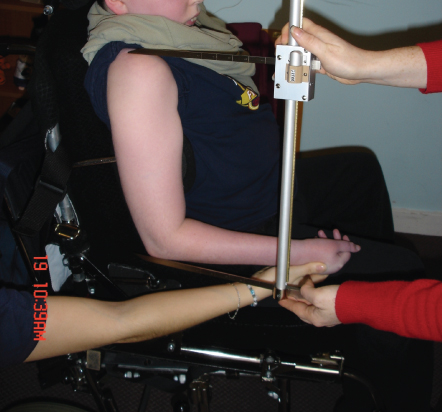
Figure 29.1 Upper arm length.
Lower leg length
The lower leg length, also known as the tibial length (TL), is measured from the tibia to the sphyrion. It requires the child to be sitting and is taken on the right side or the least affected side. This measurement can be taken accurately with an anthropometer or steel tape measure (Fig. 29.2). Two measurements should be taken and averaged. The measurement can be converted into a height measure and plotted on a growth chart, using the following formula [59]:
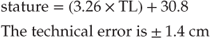
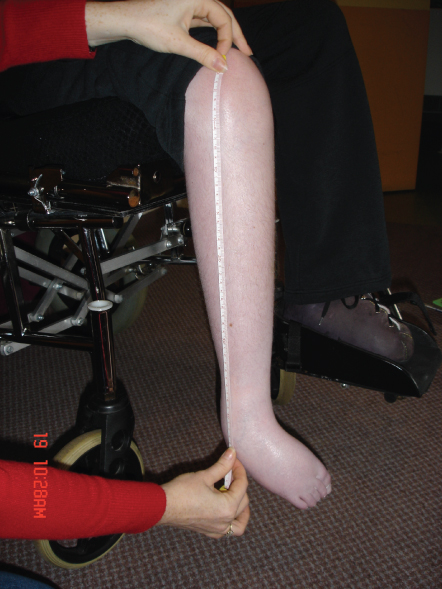
Figure 29.2 Lower leg length.
Knee height
Knee height (KH) is measured with the child sitting down and the knee and ankle bent to 90°. Using a sliding caliper the distance between the heel to the superior surface of the knee over the femoral condyle is measured on the left side or least affected side (Fig. 29.3). Two measurements should be taken and averaged. The measurement can be converted into a height measure and plotted on a growth chart, using the following formula [59]:
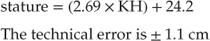
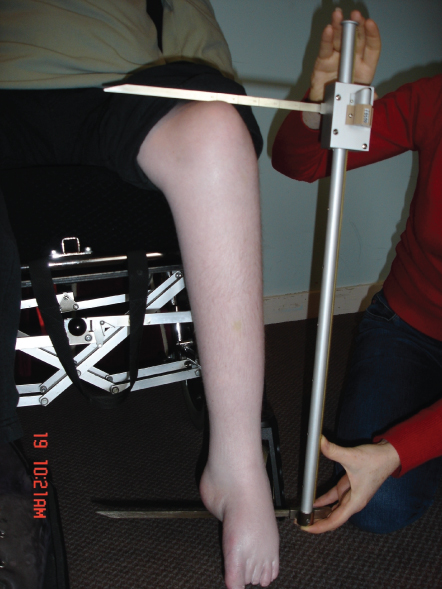
Figure 29.3 Knee height.
When an alternative length measurement is taken, note of the limb from which the measurement was taken should be made, and this should be consistently used for all subsequent measures. There are centiles specifically for each of the alternative measurements based on American data by Snyder et al. [59], but these tables are not widely available at present in the UK; thus conversion to a height measure and plotting on a growth chart is recommended.
Body composition
There is good evidence that body composition of disabled children can be ascertained by measuring skinfold thickness. Triceps and subscapular skinfold thicknesses in particular correlate highly with true fat and fat free mass. However, in routine practice, it can be difficult to take accurate skinfold thickness measurements, e.g. subscapular skinfold thickness measurement may be impractical due to the need to remove clothing or spinal jackets. In practical terms annual serial measurements of mid arm circumference (MAC) and triceps skinfold thickness (TSF) can be a useful monitoring tool and can be used to estimate mid arm muscle circumference (MAMC) [60]:
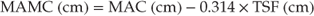
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


