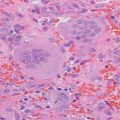
Sporadic primary hyperparathyroidism
Familial primary hyperparathyroidism
Adenoma (85 %)
Isolated
Hyperplasia (15 %)
MEN1
Carcinoma (<1 %)
MEN2A
MEN4
Hyperparathyroidism-jaw tumor
Familial hypocalciuric hypercalcemia
Tertiary hyperparathyroidism
Autoimmune hypocalciuric hypercalcemia
Chronic renal failure
Lithium-associated hypercalcemia
Thiazide-associated hypercalcemia
FHH is an autosomal dominant disorder with a nearly 100 % penetrance of hypercalcemia at all ages most commonly caused by a heterozygous inactivating mutation of the CASR gene, which encodes the calcium-sensing receptor (CaSR) [2, 3]. Homozygous inactivating mutations are found in neonatal severe hyperparathyroidism. The CaSR is a critical part of calcium homeostasis through its role in regulating PTH secretion and renal tubular reabsorption of calcium. The result of the inactivating mutation is an increase in the set point for calcium-induced suppression of PTH secretion and renal tubular calcium reabsorption. The CaSR is a typical G-protein-coupled receptor that is widely expressed both in tissues with direct involvement with calcium homeostasis such as the parathyroid glands and the kidneys but also in tissues without a role in calcium homeostasis [4].
Initially FHH was thought to be only caused by mutations in the CASR gene (FHH1 in ≈65 %). However, additional loss of function mutations has been identified in the G-alpha subunit 11 (GNA11 gene) (FHH2 in ≈15 %) [5] and the adaptor-related protein complex, sigma 1 subunit (AP2S1 gene) (FHH3 in ≈ 20 %), which encodes the adapter-protein 2 sigma subunit [6]. These mutations interfere with the normal signaling of the CaSR complex. On a clinical basis, these are indistinguishable from FHH caused by inactivation of the CaSR.
Distinguishing PHPT and FHH can be challenging, especially in the absence of a family history. Clear evidence of prior normal calcium values over time would argue against FHH and for PHPT. Individuals with FHH do not present with the classic complications seen in PHPT such as nephrolithiasis. However, serum PTH levels are elevated in 5–25 % of FHH subjects [7], and although renal calcium to creatinine clearance ratio below 0.01 is highly suggestive of FHH, there is considerable overlap in this ratio between FHH and PHPT. The risk of osteoporosis in individuals with FHH is similar to that expected in the general population, and forearm bone mineral density (frequently low in PHPT) was not found to be a reliable diagnostic tool to discriminate the two disorders [8].
Management
The findings of a low calcium/creatinine clearance ratio and a father with hypercalcemia were suggestive of FHH. The patient agreed to genetic testing of the CASR gene for mutations that cause inactivation and would suggest a diagnosis of FHH. The following heterozygous sequence change was detected: Exon, 7; DNA change, c.2243C>G; and amino acid change, p. P748R. This alteration is known to be disease associated with FHH. The inactivating mutations in the CASR gene giving rise to FHH now number over 150 and can occur anywhere in the coding region but most frequently are reported in the N-terminal extracellular domain and the signal transduction domain [4]. Variable elevations in serum calcium levels are seen with different CASR gene mutations. CASR gene testing can be falsely negative in approximately 30 % if the mutation is outside of the tested coding exons. Autoantibodies to the CaSR (autoimmune hypocalciuric hypercalcemia) have been reported to result in a similar syndrome of hypocalciuric hypercalcemia [9].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


