Familial hemophagocytic lymphohistiocytosis (FHL) is a rare heritable disorder of immune regulation that is typically characterized by sudden onset of severe systemic illness. Functional impairment or absence of 1 or more of several proteins that participate in lymphocyte cytotoxicity underlies the disease. Although FHL usually presents in infancy, age of onset is variable and dependent on genetic and environmental factors. Initial treatment consists of immune suppression, whereas definitive treatment requires hematopoietic cell transplantation.
Key points
- •
Familial hemophagocytic lymphohistiocytosis (FHL) is a rare hyperinflammatory condition that is genetically heterogeneous. FHL is typically diagnosed in infancy but may present much later in life.
- •
Approximately 70% of individuals diagnosed with FHL have mutations identified in a hemophagocytic lymphohistiocytosis (HLH)–associated gene.
- •
FHL bears resemblance to secondary forms of HLH, including Epstein-Barr virus–associated HLH and macrophage activation syndrome. The approach to initial treatment is similar.
- •
Hematopoietic cell transplantation is necessary to cure FHL. Reduced-intensity conditioning before transplantation is associated with lower risk of toxicity and higher likelihood of survival.
Introduction
Familial hemophagocytic lymphohistiocytosis (FHL) is a rare, life-threatening, inherited hyperinflammatory syndrome that may be clinically indistinguishable from secondary forms of hemophagocytic lymphohistiocytosis (HLH). The molecular basis of FHL is heterogeneous, with defects occurring in 1 or more of several proteins that participate in lymphocyte cytotoxicity. In most but not all cases of FHL, the genetic defect responsible for the disease can be identified. However, in some cases, the diagnosis of FHL is presumed from a relapsing or unremitting course or a positive family history. There is still much more to be learned about the interplay between genetics and environmental factors that underlie FHL.
Introduction
Familial hemophagocytic lymphohistiocytosis (FHL) is a rare, life-threatening, inherited hyperinflammatory syndrome that may be clinically indistinguishable from secondary forms of hemophagocytic lymphohistiocytosis (HLH). The molecular basis of FHL is heterogeneous, with defects occurring in 1 or more of several proteins that participate in lymphocyte cytotoxicity. In most but not all cases of FHL, the genetic defect responsible for the disease can be identified. However, in some cases, the diagnosis of FHL is presumed from a relapsing or unremitting course or a positive family history. There is still much more to be learned about the interplay between genetics and environmental factors that underlie FHL.
Incidence
FHL is estimated to affect approximately 1 in 50,000 live births or 0.12 to 0.15 per 100,000 children per year. The disease occurs worldwide among all races and ethnic groups. The absolute frequency of disease-causing mutations in specific HLH-associated genes varies among populations. A slight male preponderance may be attributable to the occurrence of FHL in association with X-linked lymphoproliferative disorders (XLPs).
FHL is a conditional disease in the sense that affected individuals are healthy until they encounter a trigger, such as an ordinary viral infection. Once initiated, the cycle of inflammation and the signs and symptoms that are elicited usually progress very rapidly. Secondary HLH is more prevalent than FHL; however, the true incidence of secondary HLH in children and adults remains undefined.
Familial hemophagocytic lymphohistiocytosis subtypes
FHL was first described in 1952 in 2 infant brothers and was recognized as a rare, fatal familial disease with an autosomal recessive inheritance pattern. It was not until 1999 that Stepp and colleagues discovered the association of FHL with mutations in the perforin gene, establishing the molecular basis for FHL-2. In the years since, the molecular basis for FHL-3, FLH-4, and FHL-5 has been determined. The gene responsible for FHL-1, which is linked to chromosome 9q22, remains a mystery. In addition, 2 distinct X-linked syndromes, XLP-1 and XLP-2, are typically associated with HLH and individuals with Griscelli syndrome, Chédiak-Higashi syndrome, Hermansky-Pudlak syndrome, and certain glycogen storage disorders sometimes develop HLH.
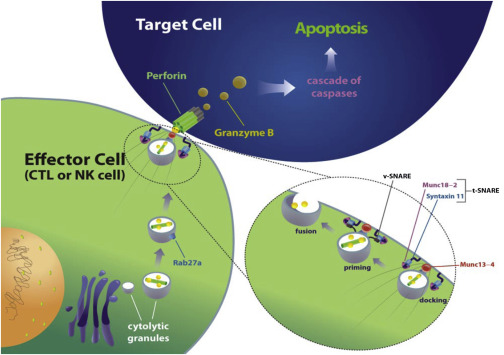
The underlying defect in patients with FHL can be ascribed to a defect in the primary cytotoxicity effector pathway in lymphocytes and natural killer (NK) cells, leading to poor clearance of target cells and a perpetual state of immune activation ( Fig. 1 ). The genetic variants of FHL are presented in Table 1 .
| Disease | Locus | Gene/Gene Symbol | Function | Syndromic Associations | Relative Frequency (%) |
|---|---|---|---|---|---|
| FHL-1 | 9q22.1-23 | Unknown | — | None | — |
| FHL-2 | 10q22 | Perforin/PRF1 | Induction of apoptosis | None | 20–30 |
| FHL-3 | 17q25 | Munc13-4/UNC13D | Vesicle priming | None | 20–30 |
| FHL-4 | 6q24 | Syntaxin-11/STX11 | Vesicle transport | None | <5 |
| FHL-5 | 19p13 | Munc18-2/STXBP2 | Vesicle trafficking and release | Colitis, bleeding tendency, hearing loss | 20 |
| GS2 | 15q21 | RAS-associated protein/RAB27A | Signal transduction | Partial albinism | — |
| CHS | 1q42.1-42.2 | Lysosomal trafficking regulator/LYST | Vesicle transport | Partial albinism, susceptibility to infection, neuropathy | — |
| XLP-1 | Xq25 | SAP/SHD2D1A | Signal transduction | Sensitivity to EBV infection, hypogammaglobulinemia, lymphoma | — |
| XLP-2 | Xq25 | XIAP/BIRC4 | Induction of apoptosis | Splenomegaly, hypogammaglobulinemia | — |
Perforin is a critical component of cytotoxic granules in effector T and NK cells. Upon effector cell degranulation, perforin and granzymes are released from the cytotoxic granules in close proximity to a target cell, such as a virally infected cell. Perforin creates a pore in the membrane of the target cell, allowing granzymes to initiate target cell death by apoptosis. Abnormal or absent perforin function leads to impaired killing of target cells and uncontrolled T-cell activation and high levels of inflammatory cytokines. This so-called cytokine storm is responsible for the tissue damage and clinical symptoms seen in HLH. Interferon (IFN) gamma is an essential player in this cascade. Blockade of IFN gamma activity using a neutralizing antibody abrogates the clinical features of HLH in perforin-deficient mice.
The median age of onset of FHL-2 is 3 months. However, there is a wide range of disease onset and severity that correlates with the degree of perforin deficiency. Residual cytotoxic function and later age of onset are observed in patients with at least 1 missense mutation. Among African Americans, FHL-2 accounts for most FHL cases. The 50delT is the most common mutation observed in this population and is associated with profound perforin deficiency and early disease onset.
The process of effector cell cytotoxicity depends on the regulated transport, docking, priming, and fusion of perforin-containing lytic granules at the effector–target cell interface. Munc13-4 participates in preparing lytic vesicles that are docked at the cell surface for fusion with the membrane. Impaired lymphocyte cytotoxicity results from Munc13-4 deficiency and biallelic mutation in Munc13-4 causes FHL-3. Disease-causing Munc13-4 mutations, which may be classified as nonsense, missense, or both, are scattered throughout the gene. Earlier age at onset is associated with biallelic missense mutation and a more severe impairment of Munc13-4 function. FHL-3 occurs in patients of all ethnicities. Although the median age of onset is 4 months, there is wide variability. Central nervous system (CNS) manifestations are especially frequent in this FHL subtype.
Mutations in the putative vesicular SNAP, syntaxin-11 (STX11), are responsible for FHL-4. STX11 associates with syntaxin-binding protein 2 (STXBP2/Munc18-2), regulating the fusion of lytic granules with the plasma membrane at the interface between effector and target cells. FHL-4 was first identified among Turkish/Kurdish families and is rare outside that population.
The binding of STX11 with STXBP2 regulates granule docking and initiation of SNARE complex formation. Mutations in STXBP2, also known as Munc18-2, are responsible for FHL-5. The age of onset of FHL-5 is highly variable and the disease is often associated with colitis, hearing loss, and abnormal bleeding.
X-linked lymphoproliferative disorders 1 and 2 are distinct X-linked syndromes characterized by susceptibility to Epstein-Barr virus (EBV) infection, HLH, and dysgammaglobulinemia. Patients with XLP-1 harbor hemizygous mutations in the Src homology 2 domain–containing gene 1A ( SH2D1A ), which functions as a lymphocyte signaling adapter. These patients have a multifactorial humoral and cellular immune deficiency that, in addition to HLH and dysgammaglobulinemia, is associated with an increased risk of lymphoid malignancy. XLP-2 has been described more recently and results from mutations in the BIRC4 gene, which encodes X-linked inhibitor of apoptosis (XIAP). Patients with XIAP deficiency may present with colitis in addition to susceptibility to EBV infection and HLH, but they do not seem to be at increased risk for malignancy.
Griscelli syndrome type 2 (GS2) and Chédiak-Higashi syndrome type 1 (CHS1) are autosomal recessive gene defects characterized by partial albinism in association with HLH. The GS2 protein, RAB27A, a small GTPase of the RAS-associated family, directly associates with the FHL-3 protein, Munc13-4, and participates in cytotoxic granule exocytosis. HLH occurs in patients with GS2 but not in patients with GS1 or GS3, which are caused by mutations in myosin-Va and melanophilin, respectively. In melanocytes, Rab27a, melanophilin, and myosin-Va are all required for distribution of pigment-containing melanosomes. However, melanophilin and myosin-Va are not required for lymphocyte cytotoxicity. Pigmentary defects may be subtle or inapparent in patients with GS2, especially those with fair skin. Like GS2, CHS is characterized by pigmentation defects, and is caused by mutations in the LYST protein, which is involved in lysosomal transport and vesicular trafficking.
Historically, FHL has been understood to be an autosomal recessive disease with high penetrance that presents in early infancy and is inevitably fatal without hematopoietic cell transplantation (HCT). However, recent and ongoing work on the molecular genetics and pathophysiology of HLH provides evidence that FHL is more varied than was once assumed. Genetic lesions in 1 or more HLH-associated genes are present in a high proportion of cases of adolescent-onset and adult-onset HLH; sequence variants in PRF1, Munc13-4, and STXBP2 were found in 25 (14%) of 175 adults with HLH who were studied retrospectively. Certain monoallelic mutations, such as one illustrative case affecting STXPB2, may be sufficient to impair lymphocyte cytotoxicity and contribute to HLH. In addition, synergistic defects in 2 (or more) distinct molecules involved in the cytotoxic pathway also can lead to HLH in some cases. Impaired cytotoxicity related to a genetic lesion in an HLH-associated gene seems to predispose to the development of macrophage activation syndrome in some patients with rheumatologic conditions.
Related posts:
 Dedication
Dedication
 Clinical Characteristics and Treatment of Langerhans Cell Histiocytosis
Clinical Characteristics and Treatment of Langerhans Cell Histiocytosis
 Strategies for the Prevention of Central Nervous System Complications in Patients with Langerhans Cell Histiocytosis
Strategies for the Prevention of Central Nervous System Complications in Patients with Langerhans Cell Histiocytosis
 Congenital and Acquired Disorders of Macrophages and Histiocytes
Familial Hemophagocytic Lymphohistiocytosis
Pathogenesis of Hemophagocytic Lymphohistiocytosis
Congenital and Acquired Disorders of Macrophages and Histiocytes
Familial Hemophagocytic Lymphohistiocytosis
Pathogenesis of Hemophagocytic Lymphohistiocytosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


