Soft tissue sarcomas comprise tumors originating from mesenchymal or connective tissue. Histologic grade is integral to prognosis. Because sarcoma management is multimodal, histologic subtype should inform optimum treatment. Appropriate biopsy and communication between surgeon and pathologist can help ensure a correct diagnosis. Treatment often involves surgical excision with wide margins and adjuvant radiotherapy. There is no consensus on what constitutes an adequate margin for histologic subtypes. An appreciation of how histology corresponds with tumor biology and surgical anatomic constraints is needed for management of this disease. Even with the surgical goal of wide resection being obtained, many patients do not outlive their disease.
Key points
- •
The early detection of disease and accurate staging at presentation are the main prognostic factors for overall survival.
- •
Among malignancies, poorly differentiated sarcoma can appear like poorly differentiated carcinoma and melanoma can mimic many sarcoma types.
- •
Communication between the pathologist, radiologist, and surgeon can expose nuance between these malignancies and be essential for final diagnosis and staging.
- •
Many advances have been made in the histologic diagnosis of STS that have, in turn, guided surgical treatment; however, even with the surgical goal of wide resection obtained in most cases, surgeons are still unable to fully cure some patients.
Introduction
Soft tissue sarcomas (STS) comprise a small group of tumors originating from mesenchymal or connective tissue. Soft tissue tumors as a whole are predominantly benign with a high rate of cure. The few that are malignant sarcomas make up less than 1% of all soft tissue tumors. All STS by definition are malignant, having the capacity to invade adjacent tissues and/or potentially metastasize to distant sites. The annual incidence of STS is estimated to be 5 per 100,000 persons, with the American Cancer Society estimating almost 12,000 new cases in 2015. The National Cancer Database places STS twenty-seventh in overall incidence in the United States, similar to Hodgkin lymphoma and cervical cancer.
STS are found across all demographics with a slight male bias (1:1.2 female/male patients), with incidence increasing after age 55 years and a median overall age of 65 years. This can be deceiving because specific sarcomas can favor younger patients, such as Ewing sarcoma and rhabdomyosarcoma existing mostly in the first 2 decades and synovial sarcoma occurring mostly in the third and fourth decades. Although comparatively rare overall, sarcomas still make up 7% of all childhood tumors.
The 5-year survival rate for all STS reported by the Surveillance, Epidemiology, and End Results (SEER) database from 2005 to 2011 is 66%, with an average length of survival after diagnosis of 7 years. The advances in overall survival made with the implementation of chemotherapy and radiation therapy have largely plateaued, and the largest prognostic factors currently are early detection and cancer stage at presentation. Metastasis alone decreases 5-year survival to 16% compared with 84% with localized disease.
Nearly 70% of STS are located in the extremities, and are 4 times more common than sarcomas of the bones and joints. Extremity sarcomas typically carry a better prognosis than tumors located in the retroperitoneum or pelvis. A superficial sarcoma, by definition, is located above the muscle fascia and most of these occur in the extremities. However, only one-third of all extremity STS are superficial.
Typical treatment of extremity STS involves surgical excision with wide margins and adjuvant radiotherapy; that is, limb-sparing multimodal therapy. This generalization overlooks the multitude of factors a surgeon must consider for planning surgical resection margins when dealing with STS. An appreciation of how histology corresponds with tumor biology and surgical anatomic constraints is needed for management of this difficult disease.
Introduction
Soft tissue sarcomas (STS) comprise a small group of tumors originating from mesenchymal or connective tissue. Soft tissue tumors as a whole are predominantly benign with a high rate of cure. The few that are malignant sarcomas make up less than 1% of all soft tissue tumors. All STS by definition are malignant, having the capacity to invade adjacent tissues and/or potentially metastasize to distant sites. The annual incidence of STS is estimated to be 5 per 100,000 persons, with the American Cancer Society estimating almost 12,000 new cases in 2015. The National Cancer Database places STS twenty-seventh in overall incidence in the United States, similar to Hodgkin lymphoma and cervical cancer.
STS are found across all demographics with a slight male bias (1:1.2 female/male patients), with incidence increasing after age 55 years and a median overall age of 65 years. This can be deceiving because specific sarcomas can favor younger patients, such as Ewing sarcoma and rhabdomyosarcoma existing mostly in the first 2 decades and synovial sarcoma occurring mostly in the third and fourth decades. Although comparatively rare overall, sarcomas still make up 7% of all childhood tumors.
The 5-year survival rate for all STS reported by the Surveillance, Epidemiology, and End Results (SEER) database from 2005 to 2011 is 66%, with an average length of survival after diagnosis of 7 years. The advances in overall survival made with the implementation of chemotherapy and radiation therapy have largely plateaued, and the largest prognostic factors currently are early detection and cancer stage at presentation. Metastasis alone decreases 5-year survival to 16% compared with 84% with localized disease.
Nearly 70% of STS are located in the extremities, and are 4 times more common than sarcomas of the bones and joints. Extremity sarcomas typically carry a better prognosis than tumors located in the retroperitoneum or pelvis. A superficial sarcoma, by definition, is located above the muscle fascia and most of these occur in the extremities. However, only one-third of all extremity STS are superficial.
Typical treatment of extremity STS involves surgical excision with wide margins and adjuvant radiotherapy; that is, limb-sparing multimodal therapy. This generalization overlooks the multitude of factors a surgeon must consider for planning surgical resection margins when dealing with STS. An appreciation of how histology corresponds with tumor biology and surgical anatomic constraints is needed for management of this difficult disease.
Histology as a prognostic factor
There is a hierarchy of prognostic factors in STS, with most converging on local recurrence, metastasis, and overall survival. The interplay can get confusing because prognostic factors vary based on endpoint. For example, in extremity STS, the prognostic factors for local recurrence are not necessarily the same as those for metastasis ; however local recurrence can itself be a prognostic factor for metastasis.
Histologic grade has an integral role in multiple aspects of STS prognosis, ranging from local recurrence to metastasis and disease specific survival as seen in Kaplan-Meier estimates. However, the wide-ranging prognostic effect of histologic grade can be underestimated in competing scenarios. This is commonly seen when interpreting the hazard ratio (HR) of histologic grade, such that the HR of one outcome can mask the effect of another. Biau and colleagues elegantly show that high-grade STS has effects on metastasis (HR = 3.47) and local recurrence (HR = 2.16). However the cumulative incidence of local recurrence of STS plateaus over 10 years, with no significant difference between high-grade and low-grade tumors. Death from metastasis precluded local recurrence in patients with high-grade STS but this should not downplay the contribution of high grade on local recurrence. A similar scenario plays out when looking at STS size and local recurrence, with local recurrence of small STS actually overtaking that of larger tumors over time.
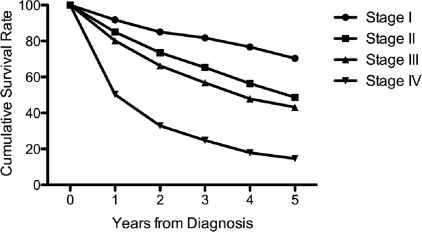
Cancer staging partly alleviates this issue by combining several prognostic factors into a single score. Cancer staging has been proven accurate and should be considered in a sarcoma patient workup. The staging system developed by the American Joint Committee on Cancer (AJCC) is most widely used. There are 4 prognostic factors that go into determining the sarcoma stage in the AJCC Cancer Staging Handbook , 7th edition, including tumor size, nodal involvement, metastasis, and histologic grade ( https://cancerstaging.org/ ). The AJCC stage is prognostic of overall survival in STS ( Fig. 1 ).
One notable change between the sixth and seventh editions of the AJCC Cancer Staging Handbook was the adoption of the French Federation of Cancer Centres Sarcoma Group (FNCLCC) histologic grading system ( Table 1 ). A unique aspect of the FNCLCC sarcoma grading system is the large contribution of a tumor differentiation score, which is given to specific STS subtypes. Some STS are given a specific differentiation score (myxoid liposarcoma is always assigned a 2), whereas others are subject to interpretation by the pathologist (angiosarcoma can receive a 1, 2, or 3). Put simply, all malignant sarcomas are not created equal. A well-differentiated leiomyosarcoma with some tumor necrosis and more than 20 mitoses per 10 high-power fields would receive a histologic grade of 2, whereas a synovial sarcoma with no necrosis and no mitotic activity would also receive a histologic grade of 2.
| Tumor Differentiation | |
| Score 1 | Sarcomas closely resembling normal adult mesenchymal tissue |
| Score 2 | Sarcomas for which histologic subtyping is certain |
| Score 3 | Undifferentiated and embryonal sarcomas, synovial sarcomas, osteosarcomas, PNET, sarcomas of doubtful type |
| Mitotic count | |
| Score 1 | 0–9 mitoses per 10 high powered fields |
| Score 2 | 10–19 mitoses per 10 high powered fields |
| Score 3 | ≥20 mitoses per 10 high powered fields |
| Tumor necrosis | |
| Score 0 | No necrosis |
| Score 1 | <50% tumor necrosis |
| Score 2 | ≥50% tumor necrosis |
| Histologic grade | |
| Grade 1 | Total score 2, 3 |
| Grade 2 | Total score 4, 5 |
| Grade 3 | Total score 6, 7, 8 |
Tumor grading was developed to be applied to STS as a whole but the histologic characteristics of specific sarcoma subtypes often carry different weights. The heterogeneity of STS can range from locally aggressive and nonmetastasizing in desmoid fibromatosis to locally indolent but highly metastatic in alveolar soft parts sarcoma. There are also a few sarcoma subtypes in which grading is not prognostic, such as malignant peripheral nerve sheath tumors.
This being said, more than 75% of all STS are high-grade (excluding benign mesenchymal tumors). This highlights the importance of obtaining an accurate histologic diagnosis in determining tumor grade in extremity STS and tumor grade should never be a substitute for histologic diagnosis.
Histologic subtypes within soft tissue sarcoma
The importance of cancer staging cannot be overstated; however, as seen when determining tumor grade, obtaining an accurate tissue diagnosis is paramount. Many different tissue types derive from mesenchymal tissue and there are currently more than 50 histologic subtypes of STS, each with unique prognostic, clinical, and therapeutic features. Because sarcoma management is multimodal, histologic subtype can and should be used to tailor optimum treatment.
Many techniques are used by the musculoskeletal pathologist when determining an STS diagnosis. In addition to traditional staining methods, such as hematoxylin and eosin, the pathologist has an array of diagnostic tools. These include ancillary immunohistochemistry (IHC), flow cytology, and several molecular techniques, such as fluorescence in situ hybridization (FISH) and reverse transcription polymerase chain reaction (RT-PCR) sequencing. There are many characteristic reciprocal chromosomal translocations unique to specific STS that can aid in histologic diagnosis ( Table 2 ). If traditional staining methods are not diagnostic, then IHC markers can help to narrow a differential diagnosis within certain tissue types ( Table 3 ). Typically a panel of stains is run to help verify tissue of origin. For histology with a broad differential an initial panel of markers might include 2 cytokeratin markers (AE1/AE3 and CAM5.2), 2 melanoma markers (S100 and Sox10), and 2 myogenic markers (SMA and HHF35). A panel of stains for just mesenchymal markers can include vimentin, factor VIII, CD31, CD34, SMA, and desmin. Sarcoma samples can also be dedifferentiated to a point at which they will not stain for characteristic markers. In these situations a multitude of factors, including tumor morphology, location, age, and medical history are taken into account by the orthopedic oncologist and pathologist when determining a histologic diagnosis.
| Translocation | Genes Involved | Sarcoma Subtype |
|---|---|---|
| t(1;3) (p36.3;q25) | Unknown | Epithelioid hemangioendothelioma |
| t(1;13) (p36;q14) | PAX7 and FHKR | Alveolar rhabdomyosarcoma |
| t(1;17) (q32;q21) | Unknown | Bizarre parosteal osteochondromatous proliferation |
| t(2;13) (q35;q14) | PAX3 and FHKR | Alveolar rhabdomyosarcoma |
| t(3;12) (q27;q14–15) | HMGA2 and LPP | Lipoma, soft tissue chondroma |
| t(7;16) (q33;p11) | FUS and CREB3L2 | Low-grade fibromyxoid sarcoma |
| t(9;15) (q22;q11–q21) | TEC/CHN and TCF12 | Extraskeletal myxoid chondrosarcoma |
| t(9;22) (q22–31;q11–q12) | TEC/CHN and EWS | Extraskeletal myxoid chondrosarcoma |
| t(11;22) (p13;q12) | WT1 and EWS | Desmoplastic small round cell tumor |
| t(11;22) (q24;q12) | FLI1 and EWS | Ewing sarcoma, PNET |
| t(12;14) (q14–15;q23–24) | HMGA2/HMGIC and various | Smooth muscle tumors, lipomas |
| t(12;15) (p13;q25) | ETV6 and NTRK3 | Infantile fibrosarcoma |
| t(12;16) (q13;p11) | CHOP and TLS | Myxoid and round cell liposarcoma |
| t(12;22) (q13;q12) | ATF1 and EWS | Clear cell sarcoma |
| t(17;22) (q21–22;q13) | COL1A1 and PDGF beta | Dermatofibrosarcoma protuberans, giant cell fibrosarcoma |
| t(X;17) (p11.2;q25) | TFE3 and ASPL | Alveolar soft parts sarcoma |
| t(X;18) (p11.2, q11.2) | SYT and SSX1/SSX2/SSX4 | Synovial sarcoma |
| Cytokeratin | Epithelial marker |
| CAM5.2 (antibody for CK8 and CK18) | Epithelial marker |
| AE1/AE3 (pancytokeratin) | Epithelial marker, SS |
| EMA (epithelial membrane antigen) | Epithelial marker, SS, melanoma |
| S100 | Melanoma and neural crest derivatives |
| Sox10 | Melanoma and neural crest derivatives |
| HMB45/Melan A | Melanoma, CCS |
| Desmin | Myogenic tumors (smooth and skeletal muscle) |
| Muscle specific actin (HHF35) | Myogenic tumors (smooth and skeletal muscle) |
| SMA | Leiomyosarcoma, myofibroblastic lesions |
| Myogenin | Immature skeletal muscle (rhabdomyosarcoma), SS |
| MyoD1 | Mature skeletal muscle |
| H-caldesmon | Smooth muscle tumors |
| CDK4 | Well-differentiated and dedifferentiated liposarcoma |
| MDM2 | Well-differentiated and dedifferentiated liposarcoma |
| CD31 | Vascular |
| CD34 | Vascular, DFSP, SFT, ES, spindle cell tumors, pleomorphic lipoma |
| ERG | Vascular, highly sensitive, and specific |
| FLI1 | Vascular, EWS |
| CD20/CD30 | lymphoma |
| CD163/CD68 | Histiocytic sarcoma |
| CD99 | EWS, SS, SFT, alveolar rhabdomyosarcoma, mesenchymal chondrosarcoma, lymphoblastic lymphoma |
| c-kit (CD117) | AML/CML, angiosarcoma, CCS, ES, EWS, melanoma, OGS, rhabdomyosarcoma |
| MUC4 | Low-grade fibromyxoid sarcoma |
As emphasized when determining tumor grade, 2 STS subtypes may look morphologically similar but have completely different behaviors. Atypical lipoma (atypical lipomatous tumor [ALT]) and well-differentiated liposarcoma are histologically identical lesions that can behave somewhat differently, with the latter having a higher rate of recurrence and risk for dedifferentiation. The difference lies in the location of the lesion that needs to be conveyed to the pathologist because a subcutaneous extremity lesion can be called an atypical lipoma or ALT, whereas well-differentiated liposarcoma is reserved for more deeply seated lesions. The goal for a surgeon is to gain not only a histologic diagnosis but an understanding within that diagnosis of the aggressive (or benign) potential of a particular specimen.
Biopsy and interpretation
Thankfully, histology gets introduced in a patient’s cancer workup relatively early. With few exceptions (ie, lipomas), nearly all extremity soft tissue masses that require surgical excision should and will have biopsies performed before excision. Histology should still factor into the surgeon’s plan when obtaining a biopsy because a representative tissue specimen needs to be delivered to the pathologist for a correct diagnosis.
The biopsy tract should be placed so it can be incorporated and excised en bloc with the definitive resection. A longitudinally oriented incision is commonly used and, for extremity lesions, a direct approach with minimal extension into adjacent tissue planes is usually possible for both superficial and deep seated lesions.
Studies have shown core needle biopsies (CNBs) are safe and less invasive than incisional biopsies. However, the accuracy of CNB in comparison with incisional biopsy for obtaining histologic diagnoses specifically in soft tissue musculoskeletal masses remains low (59.5% compared with 77.3%). Extremity STS grade determined by incisional biopsy is also predictive of both metastasis-free survival and disease-free survival, whereas grade determined by CNB is not. The main advantage of incisional biopsy is more tissue available to the pathologist to accommodate a definitive diagnosis and additional cytologic testing, and less chance of obtaining a nonrepresentative sample.
STS can grow to be quite large at presentation, with a median diameter of 5 cm for superficial and 9 cm for deep sarcomas. They are noticed incidentally on imaging or by mass effect because the initial growth for many STS is painless. The tumors tend to push adjacent structures and stay within compartments rather than invade, with the notable exception of myxofibrosarcoma (see later discussion). This means the surgeon can have ample material from which to choose a representative piece.
MRI is the modality of choice for evaluating STS. Tumor margins can be distinguished from surrounding muscle, fat, and potential neurovascular bundles. Although MRI cannot necessarily predict histologic diagnosis or tumor aggressiveness, it does provide indispensable information for biopsy planning. Areas of necrosis, liquefaction, myxoid degeneration, hemorrhage, and fibrosis are typically avoided. Abrupt changes in signal within a mass could indicate dedifferentiation and should be sampled. For many STS the most viable portions are near the periphery of the mass, where adequate nutrients for growth are present. A peripherally obtained biopsy specimen can also evaluate the extent of tumor invasion into adjacent tissues. Sampling only viable tissue can result in a falsely low grade using the FNLCC system (ie, less necrosis) and is acceptable to the alternative of an inadequate biopsy; however, it should be kept in mind the possibility that the tumor will get upgraded on final resection.
Histology interpretation does not necessarily top the list of medical skills for most surgeons, so how should one approach sarcoma histology and apply that information to interpreting a biopsy specimen? Talking directly with a musculoskeletal pathologist is ideal but is not always possible if a specimen is sent to an outside facility. Communication can be reduced to brief emails or telephone conversations, with both parties not necessarily looking at the histology slide at once. Grasping basic trends of sarcoma disease will aid in formulating pertinent questions to ask the surgical pathologist. Surgical pathology reports for different biopsy specimens can often contain the same descriptors, and only by communication can the nuances be teased out ( Table 4 ).
Related posts:
 Contemporary Management and Controversies of Sarcoma
Refinements in Sarcoma Classification in the Current 2013 World Health Organization Classification of Tumours of Soft Tissue and Bone
Contemporary Management and Controversies of Sarcoma
Refinements in Sarcoma Classification in the Current 2013 World Health Organization Classification of Tumours of Soft Tissue and Bone
 Sarcomas 2016
Management of Sarcoma Metastases to the Lung
Combined Therapy of Gastrointestinal Stromal Tumors
Myxofibrosarcoma
Sarcomas 2016
Management of Sarcoma Metastases to the Lung
Combined Therapy of Gastrointestinal Stromal Tumors
Myxofibrosarcoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


