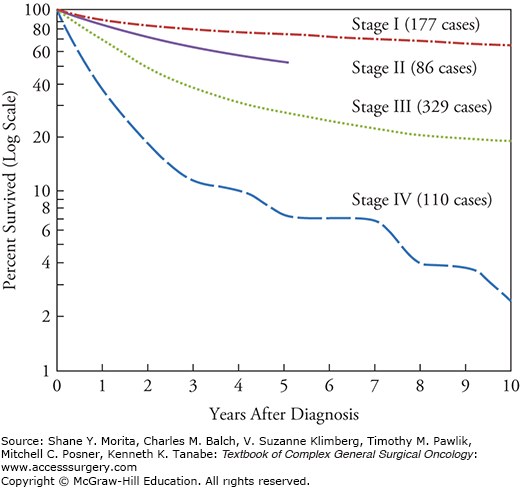
Although soft tissue sarcomas (STS) are rare, accounting for only 1% of solid tumors in adults, surgical oncologists have played a unique role performing many of the pivotal clinical trials and studies that have defined the current management and biologic behavior of these malignancies. As surgery has remained the cornerstone of treatment in patients with STS, surgical oncologists have been responsible for developing and directing their multimodality care. STS arise from or within mesenchymal tissue (fat, nerves, muscle, etc.). While early mention of possible sarcoma of nerve origin occurred in Anatomy of the Human Body by William Cheselden published in 1740,1 sarcomas were not distinguished from other cancers until the early 19th century, when Richard Virchow published his work Die krankhaften Geschwülste in 1863.2 Further pathologic classification did not occur until 1948 when Arthur Purdy Stout at Columbia University published his histogenic classification of STS subtypes.3 This allowed recognition of the differences in biologic behavior between sarcoma subtypes, however pathologic misinterpretations led to the inability to reliably diagnose sarcoma subtypes, especially when dedifferentiated or high-grade.4 Therefore, the Task Force on Soft Tissue Sarcoma of the American Joint Committee for Cancer Staging and End Results Reporting was convened, composed of pathologists and surgeons, and formulated a standardized TNM classification system in 1977 stemming from the analysis of 1215 sarcoma patient records.5 The importance of tumor grade in determining patient stage and prognosis led to the addition of grade (G) as a unique qualifier in the staging system, not folded within the tumor (T) designation (Fig. 26-1).5
Historically, treatment for STS centered solely on surgical resection. Various adjuvant therapies were attempted, with only rare reports of success. Bloodroot, a common flowering plant in North America that produces benzylisoquinoline alkaloids, was reported as a treatment for STS in the New England Journal of Medicine and Surgery in 1819.6 However, it was not until after 1963, with the FDA approval of vincristine, that alkaloid agents were formally used in the treatment of sarcoma.7 In 1891, William B. Coley published his series of nine patients, mainly with head and neck STS, that were inoculated with streptococcus erysipelas and experienced regression of disease.8 Coley went on to inoculate more than 1000 cancer patients with mainly bone and soft tissue sarcoma with bacteria or bacterial products at Memorial Hospital for the Treatment of Cancer and Allied Diseases in New York, with mixed results. Regardless, this early study into the treatment of sarcoma earned him the title of the “father of immunotherapy.”
The treatment of STS broadened with the advent of radiation therapy. STS as a group were initially considered radio-resistant, and surgical excision alone remained the preferred treatment method.9 However, starting in the 1930s, combined surgical excision with radiation therapy was performed in select cases.9 Patients who received preoperative radiation usually had bulky tumors and neoadjuvant radiation therapy was given to decrease the size of the tumor to facilitate removal or decrease the morbidity of the excision.9 Adjuvant radiation was given in surgical cases in whom margin positivity was suspected.9 A study of 653 STS patients treated at Memorial Hospital in New York found that radiation therapy combined with surgery resulted in significantly improved patient disease-specific survival in specific subsets of patients.9
Postoperative radiation therapy dramatically decreased the incidence of local recurrence in extremity STS, allowing physicians to explore limb-sparing surgical techniques. Rates of local recurrence following a radical surgical procedure had previously been as high as 25% to 50%, and amputation was the preferred surgical treatment.10 Herman Suit and colleagues10 published a landmark study out of M.D. Anderson Cancer Center in Houston in 1973 which solidified the use of radiation therapy in limb sparing surgical treatment of extremity STS. He and his colleagues found that limb-sparing surgery combined with adjuvant radiation therapy resulted in a 13% local recurrence rate at 2 years in a heterogeneous group of sarcoma patients.10 In an NCI-sponsored trial, Rosenberg et al11 prospectively randomized 43 STS patients to amputation + systemic chemotherapy versus limb sparing surgery + radiation therapy + systemic chemotherapy. No significant differences in 5-year disease-free survival rates (71% and 78%, p = 0.75) or overall survival rates (83% and 88%, p = 0.99) were detected between the limb-sparing group and the amputation treatment group.11 This trial, although underpowered, clearly demonstrated no survival benefit with amputation.
The management of STS continued to evolve with the addition of cytotoxic chemotherapies. Chemotherapy was introduced into the treatment regimen of osteogenic and soft tissue extremity sarcomas in 1948.12 However, except in the cases of childhood head and neck rhabdomyosarcoma treated with vincristine, daptomycin, and cyclophosphamide, the response was marginal.12 During the 1970s, clinical trials were initiated to study the effectiveness of chemotherapeutic drugs in the treatment of STS.11,13–16 In general, single-agent chemotherapies proved ineffective,15 however combination chemotherapy allowed for moderate improvement in patient survival.11,14 By the 1980s, 5-year overall survival of patients with extremity STS had improved to 70% to 80%.12 Unfortunately, the trials designed to assess the impact of systemic chemotherapy have generally been plagued by small recruitment numbers and have included multiple different histologic subtypes. The heterogeneity of study populations that encompass all STS patients inherently obscure any histology-specific benefit from systemic chemotherapy.
Stemming from Virchow’s original classification of sarcoma, the phylogeny of sarcoma has expanded to now to include over 60 pathologic subtypes of sarcoma (Figs. 26-2 and 26-3).17 Despite these differing subtypes, STS are still often grouped together in clinical analyses of incidence, detection, treatment, and outcomes.
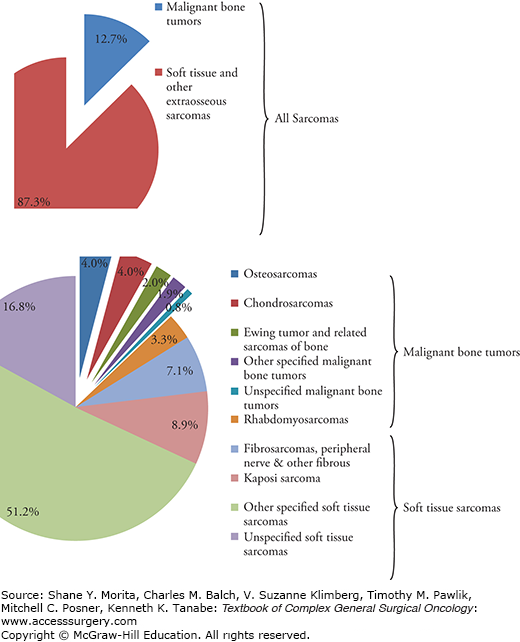
Soft tissue sarcomas account for approximately 87% of all sarcomas with bone sarcomas accounting for only 13% of all sarcoma cases (see Fig. 26-3).18 In the United States alone, it is estimated that 11,410 new soft tissue cancers were diagnosed and 4390 people died of the disease in 2013.19 International incidence rates range from 1.8 to 5.0 per 100,000 per year.20 Recent query of the Surveillance, Epidemiology, and End Results (SEER) database on all sarcoma subtypes allowed for a broader understanding of the incidence, distribution, and possible predisposing factors of sarcoma.18,21 Between 1975 and 2009, the most commonly diagnosed STS were leiomyosarcomas, Kaposi sarcomas, high-grade undifferentiated pleomorphic sarcomas (previously known as malignant fibrous histiocytomas (MFH)), liposarcomas, and fibrosarcomas.21 The incidence of sarcoma is similar by race and gender, with only slight histology-specific variations.21
Approximately 50% of STS occur in the extremities, with the trunk and retroperitoneum comprising 40% of cases, and head and neck the remaining 10%.22 The overall incidence of the various sarcoma subtypes has remained stable since the 1970s, however leiomyosarcoma and Kaposi’s sarcoma in particular have decreased in annual incidence.21,23 The decrease in Kaposi’s sarcoma of approximately 8.8% per year has been particularly profound and can partially be explained by the use of antiretroviral and antiherpesvirus therapies.23 The reported decrease in leiomyosarcoma incidence, however, is likely a reflection of the more accurate cataloging of gastrointestinal stromal tumors that were previously diagnosed as gastrointestinal leiomyosarcomas before the routine immunohistochemistry testing for C-Kit (CD117) was introduced in the early 1990s.24,25
In a study of SEER data from 2003 to 2008, the mean age at STS diagnosis was 58 years old and mean age of death from STS was 65 years old.18 Approximately 40% of STS are diagnosed after the age of 65.18 However, STS represents 8% of all childhood malignancies, with rhabdomyosarcoma accounting for half of these cases.26 Children older than five and young adults experience the lowest incidence of STS, however the incidence gradually increases until approximately age 50, at which time the rate of sarcoma development increases dramatically18.
Despite decades of study on extremity and truncal STS, few risk factors for tumor development have been identified and the etiology remains multifactorial and in some cases unclear. Variables associated with increased risk of STS include radiation exposure, viral infection, genetic predisposition and de novo genetic mutations, chemical carcinogens, chronic irritation, and lymphedema.
One of the earliest identified risk factors for STS development is ionizing radiation. In the 1920s, the first reports of radiation-induced sarcomas were published on patients treated with radiation for tuberculosis,28 and in patients with exposure to radium while painting luminescent watch dials.29 Radiation therapy in breast cancer was first associated with post-therapeutic radiation-induced sarcoma in the 1930s.30 Of the radiation-induced STS, approximately 70% are high-grade undifferentiated pleomorphic sarcomas.31 The criteria for radiation-induced sarcoma, as initially described by Cahan et al,32 include prior history of radiation therapy, latency period of several years, development of sarcoma within a previously irradiated field, and histologic confirmation of sarcoma.
Radiation therapy is often an integral part in the treatment of childhood cancer, and one of the most common secondary malignancies in these patients is STS.33 In a cohort of 4400 patients with childhood cancers followed for 20 years, Menu-Brandthomme et al33 found a 54-fold increase in the incidence of STS compared to the general population. The frequency of STS following childhood solid malignancy was 7.2-fold higher in patients initially diagnosed with Ewing’s sarcoma, which was higher than found in any other type of cancer analyzed.33 In this study, most (87%) of the secondary STS occurred within or bordering the irradiated field. The mean interval between the first cancer and the initial diagnosis of STS was 15 years (range 6 to 39 years old), and the mean age at which the diagnosis of secondary malignancy occurred was 21 years (range 9 to 46 years old).33 The risk of STS as a second malignant neoplasm was found to quadratically increase as the dose of radiation to the site of STS development increased. Generally, the risk of radiation-induced sarcoma is estimated to be up to 0.8% in exposed populations, and is dose-dependent.34
Viral infections have also been known to induce certain types of sarcoma: Kaposi’s sarcoma is the most common cancer among AIDS patients in North America and Europe, and is associated with Kaposi’s sarcoma-associated herpes virus infection. Compared to the general population in a European study, HIV-infected patients have a 128-fold increase in incidence of Kaposi’s sarcoma, and solid organ transplant recipients have a 451-fold higher incidence of Kaposi’s sarcoma.35 Antiretroviral drugs decrease the incidence of Kaposi’s sarcoma in AIDS patients.23 Epstein–Barr virus also can produce post-transplant smooth muscle tumors with microRNA expression patterns similar to leiomyosarcomas.36
There are several genetic syndromes known to be associated with the development of STS. Well-described genetic syndromes include neurofibromatosis type 1, retinoblastoma, and Li–Fraumeni syndrome. Neurofibromatosis type 1 (NF1) is an autosomal dominant disorder that occurs in 1 in every 3500 births. NF1 patients are prone to the development of benign neurofibromas and have a 10% lifetime risk of developing MPNST. NF1 patients have a mutation in the NF1 gene (17q11.2), which encodes neurofibromin, a negative regulator of the Ras pathway. Elevated Ras activity and signaling leads to increased cell proliferation.37 However, loss of NF1 is insufficient to cause MPNST, and multiple genetic aberrations contribute to the development of sarcoma in this patient population. Using genomic sequencing and copy number analysis, NF1 mutations have also been identified in myxofibrosarcoma,38 pleomorphic liposarcoma,38 and pediatric embryonal rhabdomyosarcomas.39
Retinoblastoma is a rare pediatric retinal tumor caused by a germline mutation in the RB1 tumor suppressor gene that is associated with a high risk of developing secondary malignancies due to a combination of genetic susceptibility and effects of radiation therapy.40 Soft tissue sarcomas comprise approximately 12% to 32% of secondary cancers in Rb patients, mainly leiomyosarcoma outside of radiated fields, or fibrosarcoma, rhabdomyosarcoma, and pleomorphic sarcoma localized in or near the field of radiation.40,41 Additionally, patients with familial retinoblastoma have a 500 times higher risk of osteosarcoma in their adolescence compared to the general population.42
Li–Fraumeni syndrome (LFS) is an autosomal dominant cancer syndrome characterized by the predominance of sarcoma, breast cancer, brain tumors, and adrenocortical carcinomas, often rooted in heterozygous germline mutations in the TP53 gene.43–46 The TP53 tumor suppressor gene encodes a protein involved in numerous cellular pathways regulating cell proliferation and homeostasis, such as cell cycle progression, apoptosis, and DNA repair. With TP53 genetic mutations in LFS, there is a nearly 100% lifetime cancer risk for women and greater than 70% cancer risk in men.47 Sarcomas are one of the most common cancer type seen in LFS, representing 25% of tumors in TP53 mutation carriers, with the majority (95%) occurring before 50 years of age.48,49
The landscape of STS has evolved to reflect the biomolecular foundation that distinguishes different sarcoma subtypes. Insight into the unique tumor biology illuminates targetable mechanisms of oncogenesis and disease progression and provides a platform for the development of novel molecularly targeted therapeutics.
Soft tissue sarcomas can be broadly divided into two groups: 20% fall into a category characterized by specific, balanced translocations (summarized in Table 26-1, adapted from Mertens et al50), and the remainder showing more extensive chromosomal rearrangements leading to chromosomal gains and losses.
Sarcomas with Characteristic Chromosome Aberrationsa
| Sarcoma | Aberration | Molecular Genetic Consequence |
| Atypical lipomatous tumor | Ring chromosome/giant marker | Amplification of, e.g., MDM2, CDK4, HMGA2, GLI, and SAS |
| Myxoid/round cell liposarcoma | t(12;16)(q13;p11) | FUS–DDIT3 |
| t(12;22)(q13;q12) | EWSR1–DDIT3 | |
| T(12;22;20)(q13;q12;q11) | EWS-CHOP | |
| Solitary fibrous tumor/hemangiopericytoma | der(12)(q13-15) | Unknown |
| Inflammatory myofibroblastic tumor | t(1;2)(q21;p23) | TPM3–ALK |
| inv(2)(p23q35) | ATIC–ALK | |
| t(2;2)(p23;q13) | RANBP2–ALK | |
| t(2;4)(p23;q21) | SEC31A–ALK | |
| t(2;11)(p23;p15) | CARS–ALK | |
| t(2;17)(p23;q23) | CLTC–ALK | |
| t(2;19)(p23;p13) | TPM4–ALK | |
| Infantile fibrosarcoma | t(12;15)(p13;q25) | ETV6–NTRK3 |
| Myxoinflammatory fibroblastic sarcoma | t(1;10)(p22;q24) | Deregulation of FGF8 and NPM3 |
| Ring chromosome | Amplification of, e.g., VGLL3 | |
| Low-grade fibromyxoid sarcoma | t(7;16)(q33-34;p11) | FUS–CREB3L2 |
| t(11;16)(p11;p11) | FUS–CREB3L1 | |
| Dermatofibrosarcoma protuberans | Ring chromosome, | COL1A1–PDGFB |
| t(17;22)(q21;q13), | ||
| der(22)t(17;22) | ||
| Alveolar rhabdomyosarcoma | t(1;13)(p36;q14) | PAX7–FOXO1A |
| t(2;2)(p23;q36) | PAX3–NCOA1 | |
| t(2;13)(q36;q14) | PAX3–FOXO1A | |
| t(X;2)(q13;q36) | PAX3–FOXO4 | |
| Angiomatoid fibrous histiocytoma | t(2;22)(q33;q12) | EWSR1–CREB1 |
| t(12;16)(q13;p11) | FUS–ATF1 | |
| t(12;22)(q13;q12) | EWSR1–ATF1 | |
| Synovial sarcoma | t(X;18)(p11;q11) | SS18–SSX1, SS18–SSX2 or SS18–SSX4 |
| t(X;20)(p11;q13) | SS18L1–SSX1 | |
| Clear cell sarcoma | t(2;22)(q33;q12) | EWSR1–CREB1 |
| t(12;22)(q13;q12) | EWSR1–ATF1 | |
| Desmoplastic small round cell tumor | t(11;22)(p13;q12) | EWSR1–WT1 |
| Ewing’s sarcoma family | t(11;22)(q24;q12) | EWS-FLI1 |
| t(21;22)(q22;q12) | EWS-ERG | |
| t(7;22)(p22;q12) | EWS-ETV1 | |
| Extraskeletal myxoid chondrosarcoma | t(3;9)(q12;q22) | TFG–NR4A3 |
| t(9;15)(q22;q21) | TCF12–NR4A3 | |
| t(9;17)(q22;q11) | TAF15–NR4A3 | |
| t(9;22)(q22;q12) | EWSR1–NR4A3 | |
| Alveolar soft part sarcoma | t(X;17)(p11;q25) | ASPSCR1–TFE3 |
Of the 80% of STS subtypes that show more complex genetic aberrations, clarity in the underlying biomolecular pathways is being achieved through the use of polymerase chain reaction (PCR) to detect known gene abnormalities, reverse transcriptase PCR (RT-PCR) to identify mRNA transcripts, fluorescence in situ hybridization (FISH) to detect known genetic loci, and comparative genomic hybridization (CGH) analysis to identify copy number variations.
Although many molecular pathways have been implicated in sarcoma development and growth, recent scientific advances and extensive genetic modeling have allowed clinicians a better understanding of the mechanisms contributing to sarcoma subtype-specific growth. We will highlight a few of these biomolecular pathways below.
Phosphoinositide-3-kinase (PI3K) is a critical mediator of growth factor mediated proliferation and survival. The mechanisms responsible for PI3K/AKT/mTOR activation in cancer are diverse and include activating mutations, amplification, and/or overexpression. Phosphatase and tensin homolog (PTEN) is one of the most commonly lost tumor suppressors in human cancers. Cells lacking PTEN have elevated levels of phosphatidylinositol 3,4,5-trisphosphate and AKT, with activation of the AKT/mTOR pathway which acts to prevent apoptosis and promote cell proliferation.
In sarcoma, activation of the PI3K/AKT/mTOR pathway has been implicated in Ewing’s sarcoma,51,52 desmoplastic small round cell tumor,52 MPNST,53 and synovial sarcoma.54 The pathway can be activated by genomic amplification or overexpression of receptor tyrosine kinases, such as HER2, EGFR, and IGF1R, or loss of tumor suppressors such as PTEN. Two murine models, developed independently, underscored the importance of this oncogenic signaling pathway in MPNST development. Both Gregorian et al53 and Keng et al55 engineered mice with conditional knockout of PTEN in combination with Kras activation (either directly or through NF1 mutation) to produce mice with phenotypic development of MPNST. Copy number loss of PTEN, decreased PTEN genetic expression, and PTEN methylation56,57 have been described in human MPNST samples, indicating that loss of PTEN may be a common driver of MPNST in preclinical models and human disease. Clinical trials targeting this pathway in sarcoma patients are currently underway, using agents such as ridaforolimus, temsirolimus, and other mTOR inhibitors.
P53 is a tumor suppressor often inactivated in human cancer that regulates several downstream pathways implicated in cell cycle control, apoptosis, DNA repair, and cellular senescence. MDM2 is an E3 ligase that binds and regulates P53 through a negative feedback loop in which elevated nuclear P53 activates MDM2 transcription which then blocks the P53 transactivation domain, targeting P53 for degradation. In cells that overexpress MDM2, P53 is inactivated, leading to inefficient growth arrest and stunted apoptosis. MDM2 is often amplified in well-differentiated and dedifferentiated liposarcomas.58,59 The possibility of pharmacologic MDM2 inhibition decreasing cell proliferation in MDM2-amplified liposarcoma has been studied60 and may lead to the incorporation of neoadjuvant biomarker studies in liposarcoma treatment.
Soft tissue sarcomas of the extremities and trunk are often identified by the patient as a painless mass. When symptoms do occur, they often include rapid increase in size of the mass, change in palpated density from soft to hard, functional and/or neurological deficits, pain, and swelling.61 A tumor that is clinically over 5 cm, firm, and fixed should be considered a sarcoma until proven otherwise.
The classic TNM (tumor size, nodal status, and metastasis) staging platform was introduced in the 1940s in an endeavor to guide patient treatment and clarify prognosis based on patient disease factors. In sarcoma, however, grade (designated in a 3-tier system) has been critical in the determination of patient prognosis. The American Joint Committee on Cancer (AJCC) has defined STS staging using the criteria of tumor size, nodal status, metastasis, and grade (TNMG), most recently updated in 2010 with the version 7 release. Notably, it now integrates the Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC) system for tumor grading. Despite this most recent modification, STS fit poorly into this staging system, as it was primarily designed for epithelioid malignancies. A number of important prognostic factors that predict the outcome of patients with STS are not included in this staging system and sarcoma surgical and medical oncologists have progressively moved to the use of nomograms (discussed below) as a tool to more accurately predict outcome and risk stratify individual patients with STS (Tables 26-2 to 26-5).
Primary Tumor (T)62
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Tumor diameter ≤5 cm in greatest dimension |
| T1a | Superficial to fascia |
| T1b | Deep to or involving superficial fascia |
| T2 | Tumor >5 cm in greatest dimension |
| T2a | Superficial to fascia |
| T2b | Deep to or involving superficial fascia |
Nodal Status (N)62
| NX | Regional lymph nodes cannot be assessed |
| N0 | No regional lymph node metastasis |
| N1 | Regional lymph node metastasis |
Anatomic Stage62
| Stage IA | T1a | N0 | M0 | G1, GX |
| T1b | N0 | M0 | G1, GX | |
| Stage IB | T2a | N0 | M0 | G1, GX |
| T2b | N0 | M0 | G1, GX | |
| Stage IIA | T1a | N0 | M0 | G2, G3 |
| T1b | N0 | M0 | G2, G3 | |
| Stage IIB | T2a | N0 | M0 | G2 |
| T2b | N0 | M0 | G2 | |
| Stage III | T2a, T2b | N0 | M0 | G3 |
| Any T | N1 | M0 | Any G | |
| Stage IV | Any T | Any N | M1 | Any G |
The prognosis of patients with STS varies based on a number of clinicopathologic characteristics (tumor grade, size, depth, histologic subtype, site, and patient age). To address the inherent limitations of TNMG staging, nomograms have introduced a more detailed tumor-specific method of estimating patient prognosis by incorporating multiple risk factors.63 Nomograms are outcome-based statistical models using patient-specific characteristics or treatments. They have provided a tool for clinicians to guide treatment, risk stratify patients for enrollment in clinical trials, and provide a framework for patient counseling. The landmark nomogram for STS was published by Kattan, Luong, and Brennan64 at Memorial Sloan-Kettering Cancer Center, which gave insight into expected postoperative 12-year sarcoma-specific death based on 2136 prospectively followed adult patients (see Fig. 26-4).
FIGURE 26-4:
Postoperative nomogram for 12-year sarcoma-specific death based on 2163 patients treated at Memorial Sloan-Kettering Cancer Center. (Reproduced with permission from Kattan MW, Leung DH, Brennan MF. Postoperative nomogram for 12-year sarcoma-specific death, J Clin Oncol. February 1, 2002;20(3):791–796.)

Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


