Fig. 1
A Performance of the Lymph2Cx assay using an abbreviated gene set (20 genes) compared to the “gold standard method” of previously reported GEP as well as three IHC-based algorithms [31]. B a, b show the progression-free survival (PFS) and overall survival (OS), respectively, determined by the Lymph2Cx assay. c, d show the PFS and OS determined by the “gold standard method” [31]
GEP has also proven to be useful in distinguishing a particular subgroup of DLBCL, primary mediastinal large B-cell lymphoma (PMBL). It has been found to have a unique signature that can distinguish it from ABC and GCB-DLBCL [32, 33]. PMBL showed a favorable clinical outcome comparable to GCB-DLBCL and an overlap with the gene expression signature of classical Hodgkin lymphoma cell lines. Interestingly, PMBL and Hodgkin lymphoma also share some common genetic changes such as gain or amplification of 9p24 (JAK2, PDL1, and PDL2) [32] and genomic CIITA (MHC class II transactivator) breaks [34]. Eberle et al. [35] evaluated mediastinal gray zone lymphomas (B-cell lymphoma unclassifiable intermediate between DLBCL and classical Hodgkin lymphoma) by DNA methylation analysis and also found a close relationship between gray zone lymphomas and Hodgkin lymphoma as well as PMBL; however, there were important differences seen to justify the intermediate category in the current WHO classification.
For primary cutaneous large B-cell lymphomas, GEP has been particularly helpful in supporting the distinction of primary cutaneous follicle center lymphoma (PCFCL) from primary cutaneous DLBCL, leg type (PCLBL-leg type). Both diseases can have numerous large atypical cells, but the latter has a preference for the lower extremities. PCFCL also differs from PCLBL-leg type with the latter seen in older age, frequent extracutaneous involvement, and poorer prognosis [36]. The study by Hoefnagel et al. [37] further supported the distinction of these two entities by showing PCFCL and PCLBL-leg type have a GCB- and ABC-like expression profile, respectively. The PCLBL-leg type showed deregulation of several oncogenes regulating cell cycle function, such as CMYC, PIM1, PIM2, MUM1, and OCT2. The findings of an ABC-like phenotype in PCLBL-leg type and associated poor prognosis indicate that this DLBCL is similar to that of the ABC-like DLBCL, NOS.
Primary CNS lymphoma (PCNSL) has also recently been evaluated by GEP showing downregulation of 8 genes associated with extracellular matrix, cell adhesion (FNDC1, EMCN, COL12A1), and other genes involved in mediation of cell signaling (OSMR, C4orf7, NPFFR2, TPO, and MSC) [38] when compared to peripheral blood of healthy males. This study by Sung et al. found that PCNSL shows a different expression profile compared to DLBCL, NOS with genes associated with extracellular matrix, and cell adhesion (FNDC1, EMCN, COL12A1) contributes to the pathogenesis of PCNSL. They also were able to identify increased expression of JAK-1 [38, 39], suggesting the JAK-STAT pathway may have a significant role in the pathogenesis of PCNSL.
2.2 Mantle Cell Lymphoma
Mantle cell lymphoma (MCL) is typically an aggressive disease with poor prognosis. The hallmark translocation for MCL is the t(11;14) involving the CCND1 gene. The prognosis of this disease is correlated with the deregulation of cellular proliferation [40], and more aggressive disease may demonstrate high chromosomal instability secondary to disruption to the DNA damage response [41]. Hartmann et al. [42] showed deregulation of the Hippo signaling pathway, which appears to be important in the regulation of proliferation and mitotic checkpoints. Although the majority of MCL is cyclin D1 positive, negative variants [43] have been controversial in the past, but recent studies have supported the existence of this variant, which appears to behave similarly to cyclin D1-positive forms [42] (Fig. 2). SOX11 has been found to be highly expressed in MCL and is a component of the diagnostic signature. Recently, immunostaining for SOX11 has been found to be a useful adjunct in the diagnosis of MCL, particularly for the cyclin D1-negative cases [44].
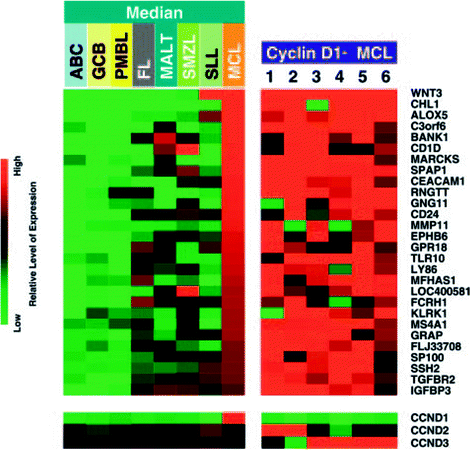
Fig. 2
Expression profiles of various B-cell lymphomas and cyclin D1-positive mantle cell lymphoma (22 cases) compared with 6 cases of cyclin D1-negative MCL. The cyclin D1-negative MCL cases showed gene expression signatures consistent with cyclin D1-positive cases [43]
Not all MCLs have aggressive disease and there are indolent forms seen as well. These patients typically have no nodal disease and present with a leukemic form, having hypermutated IGVH without complex karyotypes [41]. SOX11 protein is expressed in nearly all conventional MCLs, even cyclin D1-negative MCLs [44]. Fernandez et al. [45] suggest from their study that the lack of SOX11 protein expression in MCLs may identify a subset of patients with a more indolent behavior. These cases typically lacked p53 mutations, and ATM, or CDKN2a deletions, which are frequently seen in conventional MCLs. Recently, Liu et al. [46] looked at the tumor microenvironment of MCL and chronic lymphocytic leukemia (CLL) by GEP focusing particularly on three gene signatures (BCR, NF-κB, and MCL proliferation) and found a twofold increase in the expression of these genes in the lymph node samples compared to the peripheral blood, suggesting that MCL relies on the lymph node microenvironment for BCR engagement, cell activation, and proliferation. BCR signaling may be highly important in MCL, and this is reflected in the excellent response in patients with MCL treated with the BTK inhibitor ibrutinib.
2.3 Follicular Lymphoma
Follicular lymphoma (FL) is the second most common type of lymphoma, typically an indolent disease, and 90 % of the cases carry the classical translocation t(14;18) involving IGH/BCL2 gene. The current classification divides the tumor into grades 1–2, 3A, and 3B; however, the latter behaves more like DLBCL. GEP also supports the distinct grouping of the grades [47]. A GEP study in 2004 showed two signatures in the microenvironment, (1) immune response 1, which included T-cell-associated transcripts as well as those that were expressed in macrophages and (2) immune response 2, which were enriched in genes that were expressed in macrophages, dendritic cells, or both. Immune response 1 profile was found to be associated with a favorable survival prediction compared to the second immune response profile [48]. Recent analyses of copy number alterations (CNA) and gene mutation have provided greatly increased information on the pathogenesis and progression of FL [49–51]. This includes frequent abnormalities of genes involved in chromatin modification. One prominent example is EZH2 mutations that have been described in DLBCL as well as FL (7–22 %). Normally, EZH2 is a methyltransferase involved in repressing gene expression through trimethylation of histone 3 lysine 27 (H3K27). EZH2 mutants in lymphoma are gain-of-function mutations leading to increased trimethylation of H3K27 [52], which contributes to lymphomagenesis in germinal center B cells [53]. EZH2 mutated cases are more frequently seen in the BCL2 rearranged group and show downregulation of several genes important for differentiation beyond the GCB cell stage and TCL1A (confirmed by IHC) and upregulation of PTPN22 (Fig. 3). Inhibitors of EZH2 mutants have been developed and could have potential impacts on the management of DLBCL and FL with the mutation. A detailed discussion of all these genetic abnormalities is beyond the scope of this review, but it would be interesting to correlate GEP findings with the various genetic changes to gain a better understanding of the functional consequences associated with the alterations in future studies.
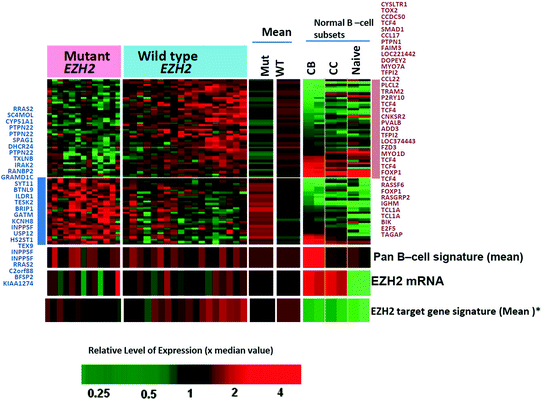
Fig. 3
Differential gene expression between EZH2 mutant versus wild-type FL cases (p < 0.01; >1.5-fold changes) [128]
Approximately 30–40 % of FLs undergo transformation to DLBCL. Although clonal relationship can be seen in paired samples of FL and transformed follicular lymphoma (tFL), currently no unifying genetic abnormalities are identified for transformation to DLBCL although some abnormalities are more common in transformed FL [49, 51]. Recurrent alterations were noted mainly in cell cycle control (MYC, CDKN2A/B), DNA damage response (TP53), immunosurveillance, and the NF-κB pathway supporting these changes are involved in various combinations in the transformation of FL [49]. Interestingly, while the majority of cases of tFL had a phenotype similar to GCB-DLBCL, transformation may be associated with a change to a more ABC-like profile. The exact mechanism of transformation requires further studies.
2.4 Burkitt Lymphoma
Burkitt lymphoma (BL) typically has a translocation of the MYC gene with one of the immunoglobulin loci, but MYC translocation is not specific for BL, and it can also be seen in DLBCL (5–10 % of cases) and other lymphomas. DLBCL typically does not have an immunoglobulin gene partner for MYC and has complex chromosomal abnormalities. The distinction between BL and DLBCL is important as the treatment is significantly different. DLBCL typically receives standard chemotherapy (R-CHOP), while BL requires intensive therapy with good response. GEP has improved the distinction between DLBCL and BL [54, 55] but not entirely as there are still cases that cannot be definitively diagnosed even with GEP. Currently, the diagnosis is based on the morphologic, immunophenotypic, and genetic findings, but there are cases that cannot be placed in one or the other category and have been designated as gray zone lymphomas (B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL). Cases that were molecularly defined BL based on high-level MYC, low-level MHC I, unique GCB signature, and low NF-κB signature, which were treated with a very intensive regimen (for BL), had an 80 % 5-year survival as compared to those with a CHOP-like regimen (20 % 5-year survival) [54]. These findings support the necessity of the distinction between these two entities for choosing the appropriate therapy. Recently, BL has been found to have a distinctive mutation landscape compared with DLBCL such as the marked prevalence of ID3 (inhibitor of DNA binding) [56, 57] and transcription factor 3 (TCF3) [58], mutations that promote cell cycle progression and proliferation. This finding may also be exploited in the diagnosis and treatment of BL. BL may arise in different settings such as endemic versus sporadic cases, HIV+ background, and EBV status that may have different genetic abnormalities and GEP.
2.5 Other B-cell Lymphomas
Marginal zone lymphoma (MZL) is a challenging diagnosis due to the lack of specific markers in many cases, heterogeneity in the morphologic features, and it is typically considered after other lymphomas (e.g., follicular, CLL) have been excluded. The distinction and interrelationship of nodal marginal zone lymphoma (NMZL), extranodal marginal zone lymphoma, MALT type (EMZL), and splenic marginal zone lymphoma (SMZL) is not entirely clear. EMZL is characterized by the API2–MALT1, BCL10–IGH, and IGH–MALT1 translocation, which commonly activates the NF-κB pathway. When gastric EMZL develops these translocations, they typically do not respond to H. pylori eradication. Increased gene expression in translocation-positive EMZLs of Toll-Like Receptors (TLR6, TLR7) and chemokine receptors (CCR2, CXCR4, CCR6, CCR7) may contribute to the persistent activation of NF-κB, leading to prolonged survival even after the obliteration of the microbe [59]. Arribas et al. [60] used GEP and miRNA expression pattern to compare SMZL with disseminated NMZL and found distinguishing patterns. Similar to a prior study [61], they found that NF-κB and CD40 pathways appear to be important in the pathogenesis of SMZL. In another recent study, Arribas et al. [62] used gene set enrichment analysis (GSEA) on 15 NMZL cases and further validated their findings on 61 paraffin-embedded NMZLs. Their study confirmed similarity of the lymphoma cells to memory B cells, as well as upregulation of genes that are important in immune response. They also found a large number of genes that are associated with NF-κB signaling pathway that are upregulated (CD74, CD81, CD82, RELA, and TRAF4). Also, the authors note that TACI (TNFRSF13B), a transmembrane activator that has multiple functions involved in apoptosis regulation and NF-κB activation, could be a new candidate gene for NMZL. However, unlike LPL and ABC DLBCL, MYD88 mutations are not commonly seen in NMZLs [63].
2.6 Peripheral T-cell Lymphomas
Peripheral T-cell lymphomas (PTCL) are relatively uncommon (10 % of all lymphomas) and often difficult to diagnose. Although there are many histologic subtypes, which include anaplastic large cell lymphoma (ALCL), angioimmunoblastic T-cell lymphoma (AITL), adult T-cell lymphoma/leukemia (ATLL), and extranodal NK/T-cell lymphoma of the nasal type, a large number of cases are placed in the not otherwise specified category (PTCL, NOS). Genetic abnormalities have been reported in PTCLs, but until recently, none have been recurrent with the exception of ALK-positive ALCLs, which is characterized by the classic t(2;5) and its variants. GEP has recently improved [64] our diagnosis and understanding of the pathobiology of PTCLs and justification of the subtypes.
Extranodal NK/T-cell lymphomas (ENKTCL) and ATLL have shown highly specific classifiers by GEP with little overlap with other entities. ENKTCLs share similar signatures with normal activated NK cells. NK and gamma–delta T-cell lymphomas within ENKTCL also have very similar gene expression profile reflecting their origin from the innate immune system [65]. ENKTCL has the classic findings of angioinvasion/angiocentricity, which can be highlighted by the gene expressed (VCAM1, CXCL9, and CXCL10) that are associated with endothelium interaction or in the pathogenesis of the vascular damage associated with EBV-positive lymphoid proliferations [66]. In a study by Huang et al. [67], they found the receptor tyrosine kinase PDGFR α was expressed at a higher level compared to normal NK cells and in vitro the ENKTCL cell line was sensitive to imatinib. Although ENKTCL may share some similarities with cytotoxic PTCLs, the prior shows greater transcript levels of cytotoxic markers, particularly granzyme H. Another study on FFPE samples showed increased expression of antiapoptotic genes as well as activation of MYC, NF–κB, and deregulation of TP53 in ENKTCLs [68]. Deletion of chromosome 6q21 appears to be characteristic of ENKTCL (40 % of the cases as well as cell lines), which may be associated with loss of function mutations and more commonly methylation of PRDM1, a tumor suppressor gene [67, 69]. Methylation of a few other genes (SHP1, p73) [70, 71], of interest has also been reported, and global methylation studies with correlation to GEP may lead to the discovery of additional tumor suppressor genes.
ATLL has a specific geographic distribution, being endemic in southwest Japan, Caribbean, Middle-East, Papua New Guinea, and South America. The disease is associated with the retrovirus, HTLV-1. HTLV-1 encodes the oncogenic protein, Tax, which dysregulates the cell cycle checkpoints as well as suppresses DNA damage repair [72]. GEP has shown a robust classifier that includes the expression of TAX and its target genes (IL2, IL2RA, IL15R, GMCSF, and TNF–α) [72]. There is also increased expression of TSLC1 (tumor suppressor in lung cancer-1) which may enhance aggregation and adhesion to endothelial cells [73]. The increase in BIRC5 (survivin) expression in ATLL may play a role in cell survival by inhibiting apoptosis and is associated with resistance to chemotherapy and poor prognosis. CD25 (TAC) tends to be highly expressed in ATLL, and attempts have been made to target this using monoclonal antibody–toxin conjugates [74–76]. More recently, CCR4 (chemokine receptor 4) expression has been reported to be frequent in ATLL [77] and may account for the frequent infiltration of the lymphoma in skin and lymph nodes. Mogamulizumab (anti-CCR4) has exhibited antitumor effect in patients with relapsed PTCL and cutaneous T-cell lymphomas [78, 79]. BIRC5 can be a potential target for therapeutics and be used in conjunction with conventional therapy for ATLL [80].
AITL has shown a prominent signature of T follicular helper cells (TFH) as well as B cell, follicular dendritic cell, and angiogenesis-related microenvironment genes. Interestingly, the prognosis of AITL patients is highly related to the stromal signature expressed [81]. Isocitrate dehydrogenase 2 (IDH2) mutations can be seen in gliomas and acute myeloid leukemia but has not been detected in other PTCLs or subtypes and appears to be fairly specific for AITL and seen in 30–40 % cases [82]. IDH enzymes normally convert isocitrate to α-ketoglutarate (α-KG); however, cancer-associated mutations result in the acquisition of new enzyme activity that converts α-KG to 2-hydroxyglutarate (2HG) which leads to DNA and histone hypermethylation through the inhibition of 2 oxoglutarate dependent dioxygenases including histone demethylases and the TET family of DNA demethylase enzymes [83]. It appears there is no prognostic implication between IDH2 mutated and non-mutated forms; however, these findings may have therapeutic implications as these tumors may be sensitive to reversing DNA hypermethylation with drugs such as 5-azacytidine or drugs specifically inhibiting IDH2 mutant activity.
ALK-negative ALCL has been included in the current 2008 WHO classification as a provisional entity although some have felt that these tumors are better classified under PTCL, NOS. IHC and morphologic findings suggest that this entity is distinct from PTCL-NOS with more similarities with the ALK+ ALCL. Earlier studies [84] by GEP have shown that ALK-negative ALCLs are closely related to ALK+ ALCLs but separated from PTCL, NOS, which has been confirmed with more recent studies [64, 84, 85]. Salaverria et al. [86] evaluated ALCLs by comparative genomic hybridization (aCGH) and found gains in 17p and 17q24-qter with losses in 4q13-q21 and 11q14 were associated more with ALK+ tumors, while 1q and 6p21 gains were more common in the ALK-negative group. Iqbal et al. [81] was able to develop a diagnostic signature for ALK-negative ALCLs that included the three genes (TNSFR8, BATF3, and TMOD1) signature reported previously. They further showed significantly increased expression of MYC and IRF4 target signatures, as well as proliferation and mTOR pathway signatures compared to PTCL, NOS [64]. ALK-negative ALCLs may benefit from aurora kinase inhibitors in conjunction with drugs that target PI3-kinase/AKT pathway [87]. The STAT3 signaling pathway [84] is activated in ALK+ ALCL and appears to be a major mechanism which promotes oncogenesis and suppresses antitumor immunity [84]. It could be a promising additional target for therapy aside from the inhibition of ALK+ [88–90]. As both ALK+ and ALK-negative ALCL highly express CD30, they are both good candidates for therapy with brentuximab vedotin [91, 92].
With GEP, many of the cases from the PTCL, NOS group can be reclassified into other established categories, but there is still a large group of cases that remain in this group. Recently, two major distinct subgroups with significantly different outcomes can be separated by GEP [64] (Fig. 4). The first group has high expression of GATA3, which appears to be associated with a poor survival compared to the other group that is TBX21 high. Within the latter group, two additional groups were identified, one with a high cytotoxic T-cell transcript expression and the other with a low transcript expression. The cytotoxic-high subgroup showed a poorer outcome compared with the cytotoxic-low subgroup. Poor clinical outcome of cases with high expression of GATA3 was confirmed by an independent study using IHC assays [93]. Patients within the GATA3 and TBX21 subgroups preferentially express different pathways and may benefit from drugs targeting some of these pathways such as mTOR inhibitors (e.g., rapamycin, temsirolimus).
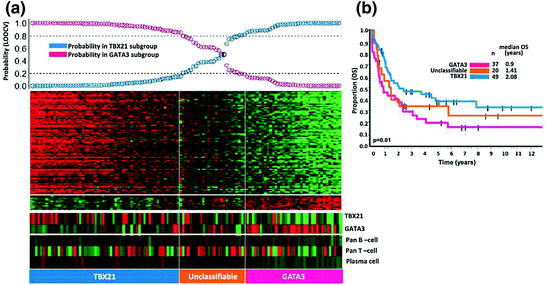
Fig. 4
Two major molecular subgroups within PTCL, NOS with biological and overall survival difference. The overall survival between the TBX21 subgroup and GATA3 subgroup was significantly different [64]
Although there has been a significant progress in the further characterization and classification of mature T-cell and NK cell lymphomas, there are still rare subtypes (e.g., many of the extranodal T-cell lymphomas) that require additional studies and collaboration from multiple large institutions will be necessary to obtain sufficient cases for meaningful studies of these rare entities.
3 Future Directions in GEP
Evaluating lymphomas by GEP has expanded our understanding of the biology and pathogenesis of these tumors, but the power of these studies will be much enhanced from the combination of GEP with additional genetic analysis, e.g., chromosome CNAs and somatic mutations as well as DNA and histone methylation status.
Both single nucleotide polymorphism arrays (SNP-A) and array comparative genomic hybridization (aCGH) have been extremely powerful in karyotypic analysis and have markedly improved resolution over conventional metaphase cytogenetics. SNP-A have advantages over aCGH and conventional cytogenetics in that it is able to detect copy number neutral loss of heterozygosity or uniparental disomy. These technologies are objective, can be automated and systematically analyzed using biostatistical algorithms [94], and have proven to be powerful discovery platforms in multiple hematologic malignancies from plasma cell myeloma [95], ALL [96], to myeloid disorders including AML [97] and MDS [98]. Aberrant DNA methylation is common in hematologic malignancies, and abnormal hypermethylation may lead to inactivation of tumor suppressor genes, while hypomethylation may lead to activation of oncogenes. Correlation of the corresponding data from GEP can be very helpful when interpreting results from these studies. GEP not only provides the expression levels of the genes of interest but also functional data of pathway activation. This information will help to identify likely candidate genes among the genes in different loci of interest [99]. It may also help to identify driver mutations, and putting mutated genes into functional pathways from a list of genes found to harbor somatic mutations. Correlative studies may not yield the desired information because of the low expression of the gene in tumor cells or expression in multiple cell types in the tumor or low tumor content. It may also be due to incomplete information in studies such as the lack of a complete picture of the methylation status of a gene [100].
While the study of protein coding transcripts has yielded highly useful information, it is now clear that noncoding RNAs are also a highly significant and important component of the transcriptome. MicroRNAs (miRNAs) are small regulatory RNAs that post-transcriptionally regulate the expression of large groups of genes that play a critical role in a broad range of biological processes [101, 102]. MiRNAs can be found in blood as well as tissue and are generally well preserved and can be extracted from FFPE tissue, which makes them useful biomarkers for investigation from archival materials including serum or plasma. MiRNAs have been found to be important in the pathogenesis of B lymphoblastic leukemias (miR-155) [103], MCL (miR-17-92) [104], CLL (miR-15a/16-1 cluster) [105], ALCL (miR-17-92), and BL (miR-28) [106]. MiR-155 has also been found to be dramatically elevated in a number of B-cell lymphomas (Hodgkin lymphoma, ABC DLBCL, PMBL, BL) [107, 108]. Overexpression of miR-155 activates the PI3K-AKT signaling pathway likely through the inhibition of p85α [109] and thus increases cell viability. Interestingly, while miR-17-92 is a MYC target, many miRNAs have been shown to be downregulated by the MYC oncogene and their repression may contribute to lymphomagenesis. A more comprehensive review is beyond the scope of this article, and the readers are referred to excellent reviews on this topic [110–112]. The role of miRNAs in lymphomas is an exciting and important discovery that will complement the data obtained through GEP of protein coding genes.
To further add to the vast information in the transcriptome is the recent discovery of long noncoding RNAs (lncRNA), which may participate in almost every step of a gene’s life cycle from transcription, splicing, decay to translation [113]. Therefore, one could imagine alterations in lncRNA expression could be highly correlated with disease. LncRNAs have been implicated in the prognosis of breast, lung, and liver cancers, but the exact role of these noncoding RNAs is yet to be defined. Preliminary data have been recently presented at national meetings and have shown exciting implications of lncRNAs in lymphomas such as MCL and Hodgkin lymphoma [114]. Coding RNA may on occasion affect the function of ncRNA by acting as a competitive inhibitor through multiple binding sites as the case of HMGA2 and the Let7 family of miRNAs [115]. With the recent advances in the area of RNA biology, GEP will be expanded to gain a better understanding of the role of noncoding RNA in lymphomagenesis and disease progression.
4 Evolution of the Platform in GEP
With the advent of next-generation sequencing (NGS), it is now possible to obtain the whole transcriptome of a tumor sample at sufficient depth for gene expression profiling and at a reasonable cost comparable to array-based GEP. Technical problems that may be associated with probe hybridization would be eliminated, but transcripts could still contain regions that are difficult to sequence. Microarrays generally lack sensitive in the range of a low copy number transcript, but with sufficient coverage, sequencing is much better in quantitating these low-level transcripts. Alternatively, spliced forms can be readily quantitated by transcriptome sequencing, and in addition, fusion transcripts and somatic mutations can be detected through sequencing technology but not by the microarray platform. For example, gene fusion involving MHC class II transactivator (CIITA) was identified in Hodgkin lymphoma and primary mediastinal B-cell lymphoma [34]. TP63 rearrangements were identified in peripheral T-cell lymphoma using RNA-seq, and further studies confirmed that the rearrangement occurred in 11 (5.8 %) of 190 cases, which was associated with inferior overall survival [116]. The expression of immunoglobulin genes, T-cell receptor genes or killer inhibitory receptors genes (KIR), and C-type lectin molecules in NK cells can also be readily detected and quantitated through transcriptome sequencing. This is useful in clonality analysis as well as in determining the repertoire of immune receptor expression in particular individuals or conditions. However, whole transcriptome sequence analysis involves a much larger data set, sequence alignment, and greater complexity in their analysis. With the development of robust and user friendly analytical programs, it is likely that more and more routine molecular biology laboratories would be able to employ this platform for their studies.
GEP relies on RNA extracted from cells or tissue samples with a varying degree of cellular heterogeneity. There is substantial interest in obtaining GEP from specific cellular populations or even single cells [117] to unravel the complexities inherent in the study of mixed populations. This can be achieved by tissue microdissection or flow sorting of disaggregated cells [118, 119]. Recently, Fluidigm has introduced a platform (BioMark HD) that uses microfluidic technology to extract mRNA from single cells and synthesizing cDNA on the same device allowing large-scale single cell GEP [120].
5 Translating Signatures to Routine Clinical Practice
One can argue that obtaining global gene expression profiling data through microarray or transcriptome sequencing would be highly valuable for personalized medicine. In a clinical trial setting, the effectiveness of pathway-directed therapy can be evaluated by correlating between pathway activation and response to the drug administered. For example, one can evaluate whether lymphomas with STAT3 pathway activation are more likely to respond to JAK1 and 2 inhibitors. If that is proven, then STAT3 activation can guide the selection of specific inhibitors in the future. In another scenario, there may be strong preliminary evidence to suggest that a drug will be effective for a certain type of lymphoma and it is desirable to perform additional validation studies, and if it is validated, then subsequent therapy can be administered according to the type of tumor present. For example, there is evidence that ibrutinib is specifically effective against the ABC type of DLBCL but not the GCB type of DLBCL. It would be important to validate this finding, and if validated, subsequent treatment should be administered to patient with the ABC but not GCB type of tumors. Accurate classification of DLBCL becomes important in the validation studies and possibly subsequent therapeutic intervention. The gold standard for the ABC versus GCB classification is the reported microarray-based GEP signature [10, 11]. There have been a number of reported IHC algorithms attempting to reproduce the GEP classification through the combination of a number of immunostains [22, 23]. Due to the difficulties in the standardization of IHC assays, it may not be an ideal assay for use across numerous institutions. It is possible to reduce the original diagnostic signature to a small set of transcripts and yet retaining the diagnostic power. Traditionally, assays such as quantitative RT-PCR have been used to quantitate these transcripts. There are several new technologies available that are highly sensitive and quantitative such as the RNA protection-based assay by high-throughput genomics or the NanoString nCounter technology.
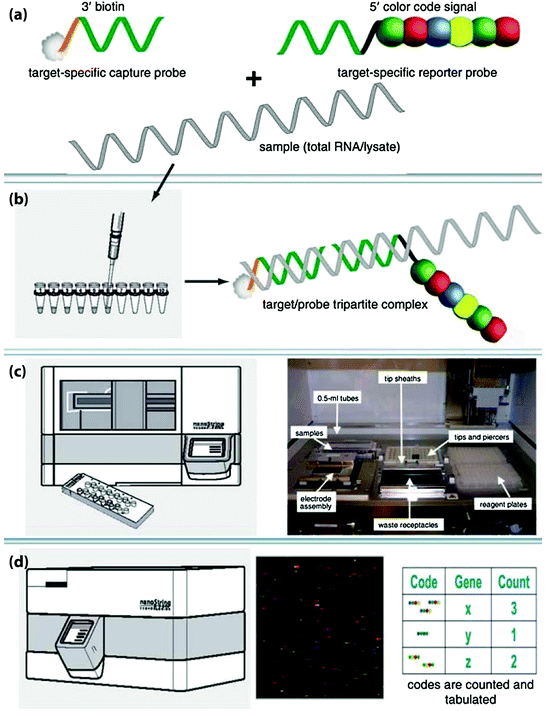
NanoString nCounter technology has been developed to capture and count specific nucleic acid molecules using color-coded probes [121]. Briefly, the technology employs a capture probe and a reporter probe per target mRNA. The former is coupled to a tag such as biotin for binding to a surface, and the latter provides the detection signal. The probes are first hybridized to the mRNA and then captured on a surface and oriented, and each color code generated from the reporter is counted (Fig. 5). The sensitivity and accuracy of nCounter are similar to that of real-time PCR but does not need any enzymatic reactions [121]. The current NanoString nCounter platform is able to measure up to 800 transcripts on one setting [122]. Payton et al. [123] studied a subtype of acute myeloid leukemia using NanoString nCounter, and the results were highly concordant with their microarray data. Several studies have evaluated whether formalin-fixed and paraffin-embedded (FFPE) samples are suitable for the NanoString nCounter technology. A study evaluated 20 genes and found a higher correlation coefficient between fresh-frozen and FFPE samples was obtained compared to quantitative real-time PCR [124]. Other studies also indicated that archival FFPE tissues can be used for retrospective transcriptome-based class prediction analysis [125]. As mentioned above, a recent report has demonstrated that a microarray-derived classifier for ABC versus GCB-DLBCL can be transferred to the NanoString platform and provide robust classification results [31]. Nuclease protection assay (NPA) is a highly reproducible and sensitive method for detection and quantitation of mRNA levels. This assay utilizes antisense probes to capture specific mRNA from the tissue, and unhybridized RNAs are digested using S1 nuclease. The captured RNA will hybridize to immobilize complementary probe at one end and linked to probes for signal detection at the other end (Fig. 6). This method also analyzes mRNA without the necessity of extraction followed by qRT-PCR [126]. Recently, several studies have used quantitative NPA to detect specific gene expression profiles in DLBCL, and the results were highly correlated with the data acquired from microarray studies [14, 127].
 Hematopoietic Cell Transplantation in Non-Hodgkin’s Lymphomas
Hematopoietic Cell Transplantation in Non-Hodgkin’s Lymphomas
 and Therapy of Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma
and Therapy of Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma
 Lymphoproliferative Disorders
Lymphoproliferative Disorders
 of Diffuse Large B-Cell Lymphoma (DLBCL)
of Diffuse Large B-Cell Lymphoma (DLBCL)
 Therapeutic Strategies and New Treatment Paradigms for Follicular Lymphoma
Therapeutic Strategies and New Treatment Paradigms for Follicular Lymphoma
 of T-Cell Lymphomas: Diagnosis and Biomarker Discovery
of T-Cell Lymphomas: Diagnosis and Biomarker Discovery




Related posts:
Hematopoietic Cell Transplantation in Non-Hodgkin’s Lymphomas
and Therapy of Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma
Lymphoproliferative Disorders
of Diffuse Large B-Cell Lymphoma (DLBCL)
Therapeutic Strategies and New Treatment Paradigms for Follicular Lymphoma
of T-Cell Lymphomas: Diagnosis and Biomarker Discovery
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
