Fig. 12.1
(a) Example of a left parietal diffuse low-grade glioma. Brain tissue specimens were sampled inside the tumor (Tu) and from the neocortex infiltrated by sparse glioma cells (Cx). Intraoperative field potential recordings demonstrated sponteanous interictal-like epileptiform discharges from the infiltrated neocortex adjacent (Cx) to the glioma core but not in the glioma core itself (Tu). (b) The proportion of patients or slices from which sponteanous interictal-like epileptiform discharges were (black) or were not (white) detected, grouped by history of seizures, and histopathological abnormalities (tumor mass vs. high tumor infiltration vs. low tumor infiltration by sparse glioma cells). (c) Western blots for Na-K-2Cl cotransporter 1 (NKCC1, left) and K-Cl cotransporter 2 (KCC2, middle) in control neocortex (control) and in neocortex close to a diffuse low-grade glioma (LGG). NKCC1 expression was normalized to a nonneuronal-specific marker, α-tubulin. KCC2 expression was normalized to a neuron-specific marker, β3 tubulin. Histogram representing NKCC1 protein quantification shows that NKCC1 is increased in the glioma sample as compared to controls. Histogram representing KCC2 protein quantification shows that KCC2 is decreased in the glioma sample as compared to controls. Representative images of KCC2 expression (red) with neuronal nuclear antigen (NeuN) marker (green). In control tissue (control), the fluorescence is distributed along the plasma membrane of the cells. In the diffuse low-grade glioma, the distribution of KCC2 in neurons in neocortex with a low tumor infiltration by sparse glioma cells is restricted in the cytoplasmic region with a loss of fluorescence along the plasma membrane of the cells. Scale bar, 5 mm
12.3.2 Intratumoral Epileptogenic Mechanisms
Since a DLGG is a space-occupying lesion, which also permeates the surrounding functional brain with infiltrating and migrating glioma cells, it may contribute to produce epileptic activities by mechanical effects [2]. Mass effect and edema may induce microcirculation impairments by reducing cerebral perfusion responsible for focal ischaemic changes in the surrounding neocortex [35]. DLGGs that grow slowly and invade the surrounding brain may isolate and deafferentate cortico-subcortical local and distant networks, leading to epileptogenicity [33, 36]. Contrarily, high-grade gliomas that grow rapidly and induce neoangiogenesis, may provoke acute tissue damages such a haemorrhage or necrosis that may participate in epileptogenesis [36]. However in DLGG, the lack of significant correlations between seizures and tumor volume, mass effect, edema, necrosis, histopathological and molecular findings and the existence of a positive correlation between seizures, cortex involvement and tumor location argue for the implication of interactions between tumor and neocortex rather than intrinsic tumor properties alone in epileptogenicity [1, 2, 33, 35].
Structural reorganization and functional deafferentation with neuronal and glial losses, neurogenesis, reactive astrogliosis, neuronal, axonal and synaptic plasticity within the peritumoral neocortex have been described, resulting mainly in a reduction of inhibitory pathways and in an increase of excitatory ones [33, 35, 36]. Accordingly, magnetoencephalography has shown that gliomas interfere with normal brain function by disrupting functional connectivity of brain networks within peritumoral and distant brain areas [33]. Taken together, these findings suggest that such changes may induce alterations in local neuronal networks, leading to imbalance between excitation and inhibition and, eventually to epileptogenicity.
Glioma cells impact the surrounding environment by recruiting non-glioma cells (astrocytes, microglia, stromal cells) that provide resources and growth advantage to facilitate tumor progression by the mean of secreted factors (cytokines, growth factors, chemokines) and of extracellular communication. Glioma cells activate and attract the neighboring microglia that, in turn, can modulate glioma biology: activated microglia enhance glioma cells migration abilities by inducing the secretion of matrix metalloproteinases and glioma cells proliferation through the EGF/Pi3K/Akt pathway.
Although rarely observed in DLGG, the pathological disruption of the blood brain barrier exposes the brain to blood serum components, such as glutamate, fibrinogen and albumin, together with the release of vascular endothelial growth factor [37]. The vascular endothelial growth factor can induce edema and its subsequent mass effect and can alter the gap-junctions permeability [37]. The extravased glutamate participate to the sustained increase of its extracellular concentration. The perivascular astrocytes may uptake albumin, whom intracellular accumulation induces a downregulation of inward—rectifying K+ (Kir 4.1) channels in astrocytes, resulting in reduced buffering of extracellular K+. Further, fibrinogen and albumin can induce a reactive astrogliosis with an impaired clearance for extracellular K+ and glutamate, leading to an increase neuronal hyperexcitability in the surrounding neuronal network. Finally, immunoglobulins themselves may be incorporated in neurons and affect their behavior.
12.3.3 Peritumoral Epileptogenic Mechanisms
Glutamate homeostasis is impaired in the peritumoral neocortex. Glioma cells lack sodium-dependant excitatory amino acid transporters 1 and 2, leading to a decrease in glutamate uptake [33, 38], and highly express the system Xc- cystine glutamate transporter, leading to a increase in glutamate release [38–41], both resulting in a poor regulation of glutamate homeostasis with highly elevated and sustained extracellular glutamate concentrations, up to 100 μm and up to tenfold higher than normal. In addition, non tumoral astrocytes and activated microglia of the peritumoral neocortex display impaired glutamate clearance abilities with reduced extracellular glutamate upload and with increased glutamate release [37]. In parallel, mutations of the isocitrate dehydrogenase genes are frequent in DLGG and they lead to conversion of isocitrate to D-2-hydroxyglutarate rather than to α-ketoglutarate [42]. Consequently, D-2-hydroxyglutarate accumulates in glioma cells and the extracellular space, where it is thought to act as a glutamate receptor agonist owing to its steric analogy to glutamate [28, 43]. In clinical practice, IDH mutations in DLGG are possibly associated with a high prevalence of epilepsy [1, 22]. Last, the glutamate extravased from blood due to blood-brain-barrier disruption participated to an increase in extracellular glutamate concentrations. Glioma epileptogenicity is thus related, in part, to an excessive glutamatergic excitatory neurotransmission. The excessive extracellular glutamate may induce seizures through the facilitation of pathological pyramidal cells synchronization [39] and experimentations on glioma-bearing mice confirmed that peritumoral neuronal hyperexcitability was attributable to glutamate release by glioma cells via the system Xc- cystine glutamate transporter [31]. In accordance, increased glutamate concentration and altered glutamate transporter expression have been shown to be associated with the presence of tumor-related seizures in patients harboring a glioma [40]. Gliomas utilize the glutamate as an “autocrine tumor growth factor” to gain a growth advantage as the released glutamate enhances glioma cells proliferation and invasion with neurotoxic, pro-invasive and proliferative effects [41]. These effects are increased by glioma cell overexpression of glutamate receptors, including metabotropic glutamate receptor (mGluR) types 2 and 3, N-Methyl-d-Aspartate (NMDA) receptors and Ca2+-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors [28]. Glutamate NMDA receptors on glioma cells increase motility of these cells [41]. NMDA receptor activation, which induces Ca2+ inflow, increases glioma cell proliferation, and AMPA receptors that predominantly lack GluR2 subunit expression, allowing a Ca2+ influx and overexpress GlutR1 subunit, resulting in an increase in glioma cells adhesion to extracellular matrix components, which have been shown to promote cell motility and invasion [39]. They also result in an activation of the glioma cell proliferation through the EGF/Pi3K/Akt pathway [44] and mitogen-activated protein kinase pathways [28]. The excessive extracellular glutamate can cause peritumoral neuronal excitotoxicity through the activation of NMDA receptors on neurons adjacent to the glioma, contributing to a glutamatergic-mediated cell death [31, 38, 39, 41], thus giving glioma cells the free space required for tumor expansion where neurons die.
GABAergic signaling is also involved both in glioma growth and epilepsy. GABA levels are higher in tissue around gliomas than in the tumor core [45]. Glioma cells express GABAA receptors that contribute to the cell volume changes required for glioma cells proliferation and migration [46]. The released extracellular glutamate downregulates neuronal and nontumoral astrocytes GABAA receptors [33, 37] and the peritumoral neocortex presents reduced GABAergic inhibitory pathways with a loss of GABAergic interneurons [32] and a reduction of inhibitory synapses within pyramidal cells [47]. These alterations result in a weakening of the GABAergic inhibitory functioning within the peritumoral neocortex [31, 33, 37, 45]. Regulation of intracellular Cl− influences neuronal responses to GABA. In healthy mature neurons, intracellular Cl− is maintained at low levels by activation of K+-Cl− transporter 2 (KCC2), which cotransports Cl− with K+ out of cells, and repression of Na+-K+-Cl− transporter (NKCC1), which cotransports Cl−, Na+ and K+ into immature neurons [28]. In this context, activation of GABA receptors causes Cl− influx that hyperpolarizes cells and inhibits neuronal activity. As in other non-glioma human focal epilepsies [48] where pathological changes in Cl− homeostasis can switch GABAergic signaling from hyperpolarizing to depolarizing, aberrant expression of KCC2 and/or NKCC1 is associated with gliomas and epileptic activity, and leads to accumulation of intracellular Cl−, which contributes both to glioma proliferation and migration and epileptic activity [28, 34, 46]. The intracellular Cl− accumulation of glioma cells, up to ~100 mM and tenfold higher than normal, is actively maintained by the NKCC1 cotransporter [49], which is expressed in glioma cells at higher ranges in peritumoral cortex of gliomas than in controls through phosphorylation mediated by WNK3, which is controlled by the EGF/Pi3K/Akt pathway [34, 50, 51]. Both glioma cell division and migration require a fast volume cell reduction, which is operated by the efflux of Cl− from Cl− channels [46, 52] coupled to K+ efflux from K+ big-conductance channels, and draw water out of the cell through aquaporines [46]. All Cl−, K+ and water channels are abnormally and highly expressed in the cytoplasmic membrane of glioma cells and are spatially restricted to the leading edge of the cell and to the invadipodia. Neurons within peritumoral cortex are affected by such Cl− dysregulation were Cl− homeostasis is disrupted in about 60% of pyramidal cells [34, 53]. This disruption is caused by upregulated expression of NKCC1 and downregulated expression of KCC2 in these neurons [34, 50, 53, 54]. Such Cl− defects are responsible for depolarizing GABAergic effects that are observed ex vivo in the cortex surrounding human gliomas and that may favor epileptogenesis in this tissues [34]. Changes in the expression of Cl− cotransporters can be triggered by brain-derived neurotrophic factor (BDNF) that is released by glioma cells and activated microglia : BDNF increases expression of NKCC1 and reduces expression of KCC2 [2, 55]. In addition, a NMDA-mediated glutamate signaling also suppresses KCC2 expression, so aberrant extracellular glutamate in gliomas could exacerbate dysregulation of Cl− levels [37].
Astrocytes are also involved in K+ homeostasis by extracellular K+ buffering through Kir4.1 channels [56]. K+ buffering is impaired in gliomas by a loss of expression of Kir4.1 channels in the plasma membrane of glioma cells that is required for cell proliferation [56]. The resulting high extracellular K+ concentration together with other local perturbations, such as the alkalization of the peritumoral neocortex [35] and the alterations of the gap-junctions functioning [33, 35] may increase the excitability of the pyramidal cells. The mTOR signalling pathways, which can be dysregulated in gliomas, result from upregulation of Pi3K/Akt signalling, and mutations in PTEN [28]. Hyperactivation of the mTOR pathway affects neuronal differentiation and migration, axonal and dendritic growth, neuronal excitability, upregulation of immature forms of the glutamatergic NMDA receptor GluN2C, and impairment of Cl− regulation [28].
12.4 Seizure Control by Oncological Treatments
Several studies have supported the beneficial role of oncological treatments (surgery, radiotherapy, chemotherapy) in seizure control of DLGG-related epilepsy. The impact of oncological treatments on seizure control are illustrated in Figs. 12.2 and 12.3.
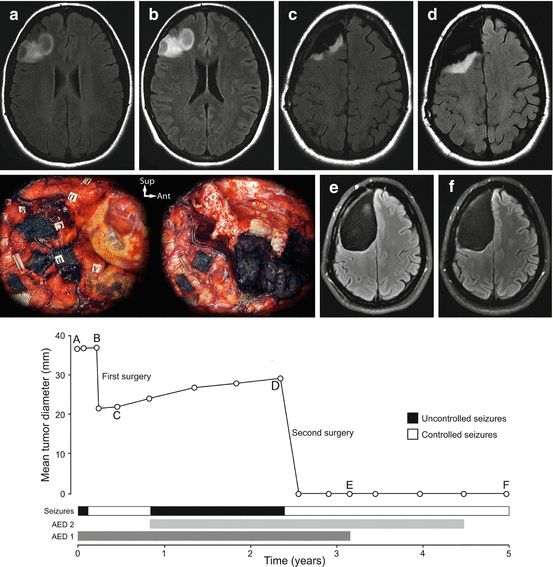
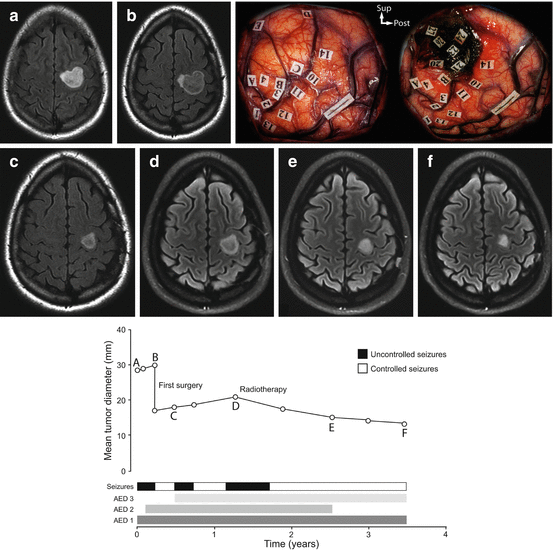
Fig. 12.2
Example of the epileptic seizure history along the natural course of a diffuse low-grade glioma. The evolution of the radiographic mean tumor diameter is plotted over time. A 33-year-old right-handed female patient presented with partial motor and secondary generalized epileptic seizures controlled with one anti-epileptic drug and a right frontal non-enhanced mass with spontaneous growth on MRI (velocity of diametric expansion at 2.6 mm/year). A subtotal resection was performed under general anaesthesia and confirmed the diagnosis of a World Health Organization grade II oligodendroglioma. 5 months after surgery, epileptic seizures recurred, requiring the introduction of antiepileptic drug 2 and the residual tumor grew on MRI (velocity of diametric expansion at 3.8 mm/year). Epileptic seizures remained uncontrolled. A second surgical resection was performed using intraoperative functional mapping with direct cortical and subcortical electrostimulations under awake condition and allowed a supratotal resection beyond MRI-defined abnormalities (see intraoperative photographs before and after resection; each numbered tag represents an eloquent site at the cortical and subcortical levels). Following this second surgery, epileptic seizures were controlled, antiepileptic drug 1 was stopped at 6 postoperative months and antiepileptic drug 2 was reduced at 24 postoperative months. At last follow-up, the patient was seizure free and no glioma recurrence was observed.
Fig. 12.3
Example of the epileptic seizure history along the natural course of a diffuse low-grade glioma. The evolution of the radiographic mean tumor diameter is plotted over time. A 26-year-old right-handed female patient presented with simple partial motor epileptic seizures that remained uncontrolled despite two anti-epileptic drugs and a left frontal non-enhanced mass with spontaneous growth on MRI (velocity of diametric expansion at 7.4 mm/year). A partial resection was performed using intraoperative functional mapping with direct cortical and subcortical electrostimulations under awake condition and confirmed the diagnosis of a World Health Organization grade II mixed glioma (see intraoperative photographs before and after resection; each numbered tag represents an eloquent site at the cortical and subcortical levels). 5 months after surgery, epileptic seizures recurred, requiring the introduction of antiepileptic drug 3 and the residual tumor grew on MRI (velocity of diametric expansion at 3.4 mm/year). At one postoperative year, epileptic seizures recurred and remained uncontrolled despite three antiepileptic drugs. A conformational external radiotherapy was performed, allowing epileptic seizure control 9 months after radiotherapy and antiepileptic drug 2 arrest 2 years after radiotherapy. At last follow-up, the patient was seizure free and no glioma recurrence was observed. Of note, radiotherapy induced a reduction of the residual glioma as quantified with a velocity of diametric expansion at −3.6 mm/year.
12.4.1 Surgery
Surgery allows seizure control in a significant proportion of diffuse DLGGs, including uncontrolled seizures. A recent systematic literature review [9] identified the extent of resection, the preoperative seizure control, and the short seizure duration before surgery as predictors of good seizure outcome after surgery. These results were reproduced in the largest studies dedicated to epileptic seizures in supratentorial DLGG in adults encompassing 1509 cases where age and extent of resection were independent predictors of postoperative seizure control while history of seizures at diagnosis, and parietal and insular locations were independent predictors of postoperative uncontrolled seizures [1]. Other predictors of postoperative uncontrolled seizures that were proposed are the presence of a preoperative neurological deficit, and the DLGG location within the central area [1, 5, 7, 9, 12, 57].
The extent of resection is the main predictor of postoperative seizure control in DLGG as illustrated in a large series of 1509 supratentorial DLGG were postoperative uncontrolled seizures occurred in 44% following biopsy, 40% following partial resection, 32% following subtotal resection, and 16% following total resection [1]. These results have improved as a result of the increased use of intraoperative mapping techniques, and awake surgery, which has made possible to increase the extent of resection according to functional boundaries while preserving the eloquent areas, but also to extend the indications for surgery within brain regions classically considered as inoperable such as the insular lobe or the rolandic area, both of these locations being highly epileptogenic [12]. It has to be noted that the intraoperative functional mapping using direct electrical cortical and subcortical stimulations under awake conditions does not increase the risk of early and late postoperative seizures [58, 59]. A major issue related to surgery, especially in paralimbic tumors, is that the epileptogenic zone can include significant extratumoral cortical areas, thus explaining why about 20% of patients still suffer from uncontrolled seizures postoperatively, even after a total tumor resection. In this setting, the removal of the putative epileptogenic foci beyond the tumor, including the hippocampal formation for paralimbic DLGG, could lead to an improved seizure outcome [9, 17]. The emerging concept of supratotal resection (i.e., removing a margin beyond MRI-defined abnormalities until functional cortico-subcortical boundaries are reached) [60] for better seizure control has been proposed [61, 62]. It relies of the presence of epileptogenic foci in the peripheral neocortex surrounding the DLGG core and infiltrated by isolated glioma cells that can be removed through a supratotal resection beyond MRI-defined abnormalities [34]. Together with the extent of resection, the short seizure duration before surgery is a main predictor of good seizure outcome after surgery [1, 7, 9, 12, 14, 63].
Second-line surgery using intraoperative functional mapping can be useful for seizure control of DLGG in case of an initial partial removal [12, 19, 64]. A series has previously demonstrated that a re-operation using intraoperative functional cortical and subcortical mapping of DLGG located within eloquent area allowed both a better DLGG control and a better seizure control [64]. In this setting, a preoperative neoadjuvant chemotherapy can be discussed and preliminary studies are encouraging [65, 66]. Last, the use of intraoperative electrocorticography monitoring may further improve postoperative seizure control by identifying surrounding epileptogenic foci [67]. However, previous series led to inconclusive results whereas it is acknowledged that the cases in which electrocorticography were used were associated with more severe and refractory epilepsy [9].
Taken together, this suggests that the main prognostic parameters of seizure control after oncological treatment of DLGG include the extent of resection.
12.4.2 Radiotherapy
Although observed in clinical practice, the impact of radiotherapy on DLGG-related seizures is supported by limited data [5, 68]. Stereotactic interstitial irradiation, which is no longer used for DLGG, allowed seizure control in 40% of patients and reduced seizure frequency in 50% of patients [69, 70]. Conventional radiotherapy has been reported to help seizure control in about 75% of DLGG patients with uncontrolled seizures [5]. A retrospective series of 33 DLGG demonstrated a reduction of 50% of more of the seizure frequency in 75% of cases following radiotherapy, 35% of patients had controlled seizures at 1 year post-radiotherapy [68]. Of note, seizure reduction usually begins early following radiotherapy and precedes the tumor shrinkage on MRI [68, 71]. The time to radiotherapy appears to impact the seizure control as the European Organisation for Research and Treatment of Cancer 22,845 phase III trial, which compared early radiotherapy versus observation and radiotherapy at “progression” in DLGG, demonstrated that 25% of patients who were irradiated had uncontrolled seizures, compared with 41% of patients who were not irradiated [72]. Of note, no difference in seizure control has been observed between high (59.4 Gy) and low (45 Gy) doses of radiotherapy [4, 73].
12.4.3 Chemotherapy
Concomitantly to the oncological efficacy, chemotherapy with alkylating agents appears to improve the seizure control of DLGG patients. Temozomide allows a reduction of the seizure frequency in 50–60% of patients, 20–40% of them being seizure free [5, 74–76]. A series of 39 DLGG demonstrated a reduction of 50% of more of the seizure frequency in 60% of patients following Temozolomide as compared to 13% in patients with antiepileptic drug therapy only [77]. A series of insular DLGG demonstrated a reduction of the seizure frequency in 100% of patients following Temozolomide, 14% of them being seizure free, as compared to 30% in patients with antiepileptic drug therapy only [19]. In this line, the emerging experience of preoperative neoadjuvant chemotherapy demonstrated a reduction of the seizure frequency in 90% of cases following Temozolomide, 50% of them being seizure free [65, 66]. Interestingly, following surgery and neoadjuvant Temozolomide, a reduction of the seizure frequency was observed in 100% of cases, 70% of them being seizure free [65]. Of note, no significant correlation between seizure response and 1p/19q codeletion has been reported so far [5].
PCV chemotherapy (procarbazine + CCNU + vincristine) allows a reduction of the seizure frequency in up to 100% of patients, up to 60% of them being seizure free [78]. A series of 33 DLGG demonstrated a reduction of the seizure frequency in 53% and a total seizure control in 31% of patients following PCV chemotherapy [79].
As a whole, alkylating agents appear as a promising therapeutic option to help the seizure control of DLGG in addition to the oncological impact, and possibly as a neoadjuvant treatment to improve the onco-functional balance of the surgical resection.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


