Fig. 20.1
The mechanism of histone acetylation and deacetylation. The modifications of histones are able to regulate gene transcription. Histone acetylase (HAT) adds acetyl groups onto the lysine tail of histones, leading to the open structure that activates the gene transcription, such as p21 and p27. Histone deacetylase (HDAC), however, removes acetyl groups resulting in compact structures to repress gene expression. HDAC inhibitors, vorinostat, TSA, panobinostat, entinostat, and romidepsin, inhibit HDAC activity, allowing gene expression to induce apoptosis. Ac acetyl group, HAT histone acetylase, HDAC histone deacetylase
As stated, deacetylation of histones gives rise to compact chromatin structure to prevent gene expression, including tumor suppressor genes, p21 and p53, leading to uncontrolled cell growth [1]. HDAC1 overexpression, for example, has been demonstrated in castration-resistant disease. HDAC1 up-regulation not only facilitates cell proliferation, but also induces the loss of luminal epithelial cytokeratin 18 (CK18), with concurrent up-regulation of cytokeratin 5 (CK5), a marker of progenitor basal cells [3] (Table 20.1). Additionally, a recent study concluded that the knockdown of HDAC1 or HDAC3 resulted in suppression of AR-regulated genes (ARG), such as KLK2, PSA, and NKX3.1, but not TMPRSS2, suggesting that HDAC1 and HDAC3 are required for ARG expression [4]. Another class I HDAC, HDAC2, was investigated in a cohort of 192 prostate cancer patients that high expression of HDAC2 was correlated with shorter relapse-free survival [5]. Welsbie et al. showed that HDAC2 does not regulate ARG expression, suggesting that the role of HDAC2 in prostate cancer development may not involve AR transcriptional activity. Moreover, the activity of HDAC 1 and 2 is positively correlated with Gleason score, indicating that HDAC1 and HDAC2 could be potential prognostic markers in prostate cancer [5]. HDAC4 is an androgen-regulated class II HDAC. Its nuclear translocation is induced by ligands in benign prostatic hyperplasia and primary prostate cancer. In CRPC, however, it is predominately located in the nuclear compartment, to drive aggressive disease progression by inhibiting differentiation [6]. HDAC6 is the major HDAC to govern the activity of molecular chaperone Heat Shock Protein 90 (Hsp90). A critical role of Hsp90 in PCa cells is to mediate AR nuclear translocation and transcriptional activity. Ai et al. demonstrated that HDAC6 knockdown prevents AR nuclear localization through acetylating Hsp90, suggesting that HDAC6 plays an important role in AR hypersensitivity in CRPC [7]. Sirtuin 1 (SIRT1), a class III HDAC, has been exhibited in association with cell proliferation and chemo resistance in PC3 and DU145. Kojima et al. demonstrated that suppressing SIRT1 in PC3 and DU145 is able to repress cell proliferation and enhance the sensitivity to camptothecin and cisplatin [8]. Other HDACs and their involvement in CRPC remain unclear. Ongoing studies within prostate cancer and other malignancies will allow us to eventually fully elucidate HDAC involvement in CPRC, and how this knowledge can be applied to novel therapeutic strategies involving HDAC inhibition.
Table 20.1
The list of HDACs in prostate cancer
Class | Members | Localization | Status in prostate cancer |
|---|---|---|---|
Class I | HDAC1 | Nucleus | Overexpression |
HDAC2 | Nucleus | Overexpression | |
HDAC3 | Nucleus | Overexpression | |
HDAC8 | Nucleus | Unclear | |
Class II | HDAC4 | Nucleus/cytoplasm | Overexpression |
HDAC5 | Nucleus/cytoplasm | Unclear | |
HDAC6 | Cytoplasm | Induce AR hypersensitivity | |
HDAC7 | Nucleus/cytoplasm | Unclear | |
HDAC9 | Nucleus/cytoplasm | Unclear | |
HDAC10 | Cytoplasm | Unclear | |
Class III | SIRT1 | Nucleus | Suppress PC3 and DU145 cell growth |
SIRT2 | Cytoplasm | Unclear | |
SIRT3 | Nucleus/mitochondria | Unclear | |
SIRT4 | Mitochondria | Unclear | |
SIRT5 | Mitochondria | Unclear | |
SIRT6 | Nucleus | Unclear | |
SIRT7 | Nucleus | Unclear | |
Class IV | HDAC11 | Nucleus/cytoplasm | Unclear |
Histone Deacetylase Inhibitors (HDACi) in CRPC
Several agents have been developed to target HDACs. These inhibitors exhibit pleiotropic effects including cell cycle arrest, DNA damage, autophagy, and apoptosis [9–11]. To date, there are at least 18 HDAC inhibitors (HDACi), divided into seven classes, being investigated in prostate cell lines or animal models [1]. In particular, vorinostat, entinostat, and panobinostat are being investigated in clinical trials.
Vorinostat, also named as suberoylanilide hydroxamic acid (SAHA), is a hydroxamate compound inhibiting class -I, -II, and -IV HDAC and has been approved to treat cutaneous T cell lymphoma by FDA in 2006 [1]. In prostate cancer therapy, it is able to induce cell death in vitro in LNCaP, LAPC4, CWR22, DU145 [12], and reduce CWR22 tumor burden in vivo [13]. Molecular analysis shows that vorinostat represses AR, PSA, and KLK2 transcription, but does not affect AR degradation [14]. Moreover, numerous studies displayed that vorinostat arrests cell cycle in G2/M, repressing EGFR expression, and has been reported to have synergistic effect with bicalutamide in DU145 [14].
Trichostatin A (TSA) is an antifungal antibiotic, specifically blocking mammalian class I and II HDACs [1]. One study showed that TSA induces cell death in LNCaP and CWR22R, but not in PC3 and DU145 [15]. Suenaga et al. suggested that TSA is capable of suppressing telomerase reverse transcriptase (hTERT) mRNA, which is responsible for maintaining telomere integrity, to restrain LNCaP and PC3 cells proliferation [16].
Panobinostat is a derivative of hydroxamic acid, the same class as SAHA. It has great activity against class -I, -II, and -IV HDAC at nanomolar ranges [1]. Some studies have indicated that panobinostat induces cell apoptosis in LNCaP [4], MYC-CaP/AS [17], MYC-CaP/CR [17], as well as PC3-AR expressing cells [18] and decreases tumor size of MYC-CaP [17], CWR22RV1 [4], PC3-AR [18] in vivo models. Several research teams have revealed that panobinostat blocks AR transcriptional activity, induces Caspase-3 activations, and attenuates ATM–Akt–ERK DNA damage pathway to cause cell death [4, 17, 18].
Romidepsin, also named as FK228, belongs to the class of bicyclic and capable of releasing a zinc-binding thiol in a cell to repress HDAC activity [1]. Sasakawa et al. have indicated that romidepsin inhibits PC3 and DU145 cell growth as well as tumor burden by inducing p21 and downregulating c-Myc expression [19]. Besides, the other group displayed that romidepsin can repress tumor volume in the 22Rv1 model and prolong survival rate [20].
Entinostat is a benzamide-based HDACi and targeting class I HDAC exclusively. It has been approved by FDA for ER-positive breast cancer treatment and investigated in LNCaP, PC3, as well as DU145 cells showing anti-tumor effect. [21]. Moreover, a study concluded that entinostat acts as a radiosensitizer to enhance radiation mediated DNA double strand breaks (DSB), as indicated by increased levels of gamma-H2AX [22]. Importantly, Qian et al. demonstrated that entinostat significantly prevents the progression of prostate tumorigenesis in TRAMP model [21].
Combination Strategies with HDAC Inhibitors in CRPC
The efficacy of HDAC inhibitors as single agents has been demonstrated in various prostate cancer cell lines and in vivo models. However, the development of CRPC results from multiple signaling pathways [24] (Table 20.2). For this reason, combining HDACi with other agents has been studied in preclinical PCa animal models and clinical trials in patients with PCa. Marrocco et al. combined vorinostat with the AR antagonist, bicalutamide, showing synergistic cell killing effect in the LNCaP cell line [14]. Also, Roklhin et al. displayed the synergistic effect of TSA and doxorubicin in LNCaP and CWR22R cells [15]. Moreover, due to the fact that panobinostat results in PI3K–AKT–mTOR pathway repression, Ellis et al. adopted Myc-CaP and PC3 models to demonstrate the greater therapeutic efficacy in vitro and in vivo by combining panobinostat with the specific mTORC1 inhibitor everolimus and the PI3K–mTOR dual inhibitor BEZ235 [17, 18].
Table 20.2
The list of the main HDAC inhibitors in vitro studies
Name of HDAC inhibitors | Combined treatment | Cell lines/animal models | Conclusions | Reference |
|---|---|---|---|---|
Vorinostat | RMT5625; HA14-1 | LNCaP; DU145; LAPC4; PC3 | Induce cell death in LNCaP and DU145, but not in PC3 | [12] |
CWR22 xenograft | Reduce tumor burden | [4] | ||
Bicalutamide | LNCaP; PC3 | The combination increases cell death in LNCaP, but not in PC3 | [14] | |
TSA | Doxorubicin | LNCaP; CWR22; PC3; DU145 | The combination enhance cell death in LNCaP and CWR22 | [15] |
LNCaP; PC3 | Increase p53 acetylation | [23] | ||
LNCaP; PC3 | Downregulate telomerase reverse transcriptase | [16] | ||
Panobinostat | Everolimus | MYC-CaP | Concurrent treatment increase cell apoptosis and inhibit tumor growth and AR transcriptional activity that this study also inhibited HIF-1 alpha transcription activity | [17] |
BEZ235 | PC3; PC3AR | Suppress tumor growth in both PC3 and PC3-AR xenografts by inhibiting AKT–mTOR–ERK pathway increases double strand breaks of DNA increases inhibition of ATM | [18] | |
CWR22Rv1 | Inhibit tumor growth | [4] | ||
Romidepsin | PC3; DU145 | Repress tumor burden in PC3 and DU145 by increasing p21 expression and decreasing c-Myc | [19] | |
CWR22Rv1 | Prolong survival rate | [20] | ||
Entinostat | LNCaP; PC3; DU145; TRAMP | Suppress tumor growth of LNCaP, DU145 and PC3. Prevent tumor progression in TRAMP | [21] | |
Radiation | DU145 | Enhance the radiosensitivity in vivo | [22] |
The promising results from these in vitro and preclinical in vivo studies involving HDAC inhibitors in various PCa models are encouraging. These results have led to numerous clinical trials, investigating the potential of HDAC inhibitors as single agent or in combination with radiotherapy, bicalutamide, docetaxel, doxorubicin, and temsirolimus [25] (Table 20.3). An example of a promising clinical trial involved the treatment of patients with docetaxel and panobinostat demonstrating PSA reduction [26]. Although most trials are still ongoing and vorinostat single treatment in CRPC has been reported high toxicity, low dose of HDAC inhibitors or combining with other drugs may still provide an avenue to treat CRPC patients.
Table 20.3
The list of HDAC inhibitor in clinical trials
Name of HDAC inhibitors | Combined therapy | Phase | Status | Dose/schedule | Conclusion | Reference |
|---|---|---|---|---|---|---|
Vorinostat (SAHA) | II | Completed | 400 mg/day, orally | 2/27 patients stable disease; significant toxicities and limiting efficacy | [27] | |
I | Completed | Dose escalation study | Unpublished | NCT00005634 | ||
I | Completed | Dose escalation study | Unpublished | NCT00045006 | ||
Docetaxel | I | Terminated | SAHA: 100-500 mg/day, orally; Docetaxel: 50-75 mg/m2 | Excessive toxicity | [28] | |
Temsirolimus | I | Ongoing | 400 mg qd | N/A | NCT01174199 | |
Doxorubicin | I | Completed | SAHA: 400-1000 mg/day; Doxorubicin: 20 mg/m2 | 1/2 patients partial response | [29] | |
Panobinostat (LBH589) | II | Completed | Unpublished | NCT00667862 | ||
Radiotherapy | I | Completed | Unpublished | NCT00670553 | ||
Docetaxel | I | Completed | LBH589: 15 mg/MWF, orally; Docetaxel: 75 mg/m2 | 2/7 patients partial response with a 50 % decline in PSA; 4/7 patients stable disease | [26] | |
Bicalutamide | I/II | Ongoing | LBH589: 60 or 120 mg/week, orally; Bicalutamide: 50 mg/day, orally | N/A | NCT00878436 | |
Entinostat (MS-275 | 13-cis retinoic acid (CRA) | I | Completed | MS-275: 4 mg/m2 weekly; CRA: 1 mg/kg/day, orally | Well tolerated | [30] |
Methylation in CRPC
Like acetylation and deacetylation of histones, methylation of DNA and histones are reversible alterations that lead to stable inheritance of cellular phenotypes without any changes in the DNA sequence (Fig. 20.2). During DNA methylation process, the methyl group (–CH3) is supplied by S-adenosylmethionine (SAM) and transferred to the 5′-carbon of a cytosine base of a CpG dinucleotide. Regions characterized by high frequency of CpG sites are defined as CpG islands and their methylation acts as a stable tag on gene promoters, leading to the formation of large-scale heterochromatic structures that silence the associated genes.
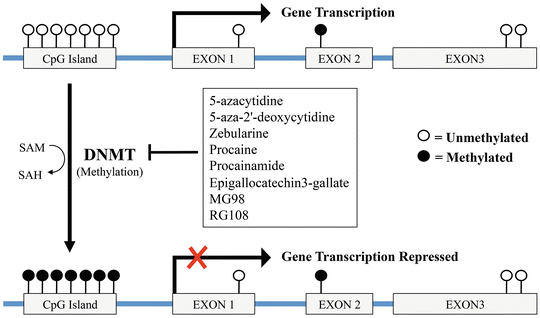
Fig. 20.2
Schematic representation of DNA methylation and its regulatory effect on gene transcription. During DNA methylation, DNA methyltransferases (DNMT) catalyze the addition of a methyl group (–CH3) to a cytosine base within a CpG island. Hypermethylation of CpG islands located in the promoter region of a gene lead to its transcriptional silencing. DNMT inhibitors, restoring the normal methylation pattern, induce the transcription of specific genes involved in apoptosis, cytostasis, and differentiation. DNMT DNA methyltransferase, SAM S-adenosyl methionine, SAH S-adenosyl homocysteine
Histone methylation occurs on arginine and lysine residues of histone tails. The methylation is catalyzed by histone methyltransferase enzymes and a single lysine residue can be methylated up to three times leading to either gene repression and activation. Both DNA and histone methylation can drive to genetic instability and alteration in normal gene transcription and have been implicated in a variety of human cancers, including PCa [31]. For those reasons, these epigenetic modifications can potentially be used for the molecular classification, detection, risk assessment, and treatment in prostate cancer. In the following sections, the principal epigenetic molecules involved in PCa progression and their relative inhibitors will be presented.
Role of DNA Methyltransferases (DNMTs) in CRPC
The DNA methyltransferases (DNMT) are a family of enzymes responsible for the establishment and differentiation of DNA methylation patterns during development. The components of this family are: DNMT1, DNMT3a, and DNMT3b. DNMT1 is considered to be the key maintenance enzyme, acting on hemi-methylated DNA substrates generated during DNA synthesis, to maintain CpG methylation patterns through genome replication and mitosis. In contrast, DNMT3a and DNMT3b perform de novo methylation. In human cancer cells, DNMT1 is responsible for both de novo and maintenance methylation of tumor suppressor genes leading to their silencing. Progressive increase in generalized DNMT enzymatic activity has been shown during malignant transformation. Gravina et al. showed that human PCa cells continuously treated with bicalutamide (BCLT) or cultured in androgen-depleted medium progressively acquire higher DNA methyltransferase (DNMT) activity and expression, mainly DNMT3a and DNMT3b, proportionally to their androgen independence [32]. These observations have also been correlated in patients, where DNMT3a and DNMT3b expression was upregulated by neoadjuvant treatment with BCLT [32]. Those results demonstrated that BCLT treatment can mediate changes in DNA methylation status thus suggesting a possible involvement in preventing the action of antiandrogen therapy. Furthermore, increased DNMT expression and activity also correlates with the up-regulation of truncated AR isoforms, which favor the development of the hormone-resistant phenotype [33].
DNA Methyltransferases (DNMTs) Inhibitors
DNMTs inhibitors represent a promising class of epigenetic modulators, showing efficient anti-tumorigenic activity in vitro and in vivo against several hematologic and solid tumors. DNMTs inhibitors are subdivided into nucleoside and non-nucleoside inhibitors (Table 20.4). Nucleoside inhibitors become incorporated into the DNA during replication and lead to the sequester of DNMTs. Non-nucleoside inhibitors do not require incorporation into DNA and are thought to be safer due to their lower mutagenic potential. Belonging to the nucleoside inhibitors are the 5-azacytidine (Vidaza) and the 5-aza-2′-deoxycytidine (or decitabine, Dacogen). 5-aza-2′-deoxycytidine is one of the first DNMT inhibitors identified. This agent forms irreversible covalent bonds with DNMT1 after its incorporation into DNA, thereby inducing degradation of DNMT1. Although different studies support the benefit of 5-aza or 5-aza-2′-deoxycytidine treatment in androgen-independent PCa cell lines [34], in CRPC patients, 5-aza-2′-deoxycytidine has been evaluated in phase II trials, but its antitumor effects resulted to be modest. Overall, 5-aza-2′-deoxycytidine led to stable disease in 17 % of patients, and median time to progression was only 10 weeks [35]. The same poor efficacy in chemonaive CRPC patients has been observed for 5-azacytidine (median progression-free survival of 12.4 weeks) [36]. Zebularine, another cytidine analog, has shown minimal acute toxic effects and higher chemical stability when compared to 5-aza-2′-deoxycytidine. Zebularine has been evaluated in preclinical studies for myeloid malignancies and selected carcinomas [37–40], but it has not yet been tested for the treatment of PCa. Non-nucleoside DNMT inhibitors like procaine and procainamide reduce DNMT affinity for both DNA and S-adenosyl-methionine causing a decrease in DNA methylation. To date, non-nucleoside analogs have been much less efficacious in inhibiting DNMT and have not been evaluated in clinical trials. However, epigallocatechin3-gallate (EGCG3), the major phenol in green tea, has been found to be a potent inhibitor of S-adenosylmethionine-dependent methyltransferase (such as DNMTs). The mechanism of its cancer-preventive action in prostate cancer is still unclear. However, a placebo-controlled, double-blind phase II trial has been conducted evaluating its ability to prevent progression towards invasive carcinoma in patients with high grade PIN lesion but no data are yet available [41]. Finally, MG98 and RG108, antisense oligodeoxynucleotides that target the 3′ UTR of DNMT1 and a small molecule able to bind to its catalytic site respectively, have demonstrated good in vitro anticancer effects with, however, a limited clinical success. Despite the promising anticancer activity in hematological malignancies, early clinical trials involving patients with solid tumors including PCa demonstrated that DNMT inhibitors have low anticancer activity and significant toxicity as monotherapy. However, recent studies suggest that low concentrations of DNMT inhibitors such as 5-Aza and decitabine may act synergistically when combined with chemotherapy and contribute to overcoming intrinsic or acquired chemoresistance in several cancer types [42].
Table 20.4
List of the main DNMT inhibitors tested and their mechanism of action (adapted from Gravina GL et al. [42])
DNMT inhibitor | Mechanism of action |
|---|---|
Nucleoside | |
5-Azacytidine | Inhibit mRNA translation and when incorporated into DNA inhibits methylation by trapping DNMTs |
5-Aza-2′-deoxycytidine/decitabine | When incorporated into DNA inhibits methylation by trapping DNMTs |
Zebularine | When incorporated into DNA inhibits methylation by trapping DNMTs |
Non-nucleoside | |
Procaine | Binds to CpG-rich sequences and block the binding of DNA methyltransferases to DNA |
Procainamide | Reduces DNMT1’s affinity for both DNA and S-adenosyl-methionine |
Epigallocatechin3-gallate (EGCG3) | |
MG98 | This antisense oligonucleotide targets the 3 UTR of DNMT1 |
RG108 | Binds to the catalytic site of DNMTs |
Role of Histone Methyltransferases (HMTs) in CRPC
Histones are the main structural components of chromosomes. These proteins assemble to form octamer and act as spools around which DNA is wrapped to form a nucleosome. Histones undergo a sophisticated pattern of posttranslational modifications, the histone code, that alter their interaction with DNA and nuclear proteins and are needed to regulate gene transcription. Histone methylation can affect chromatin remodeling and function depending on the specific amino acid being modified and the extent of methylation and can lead to both activation and repression (Fig. 20.3) [31]. The two histone methylation marks, therefore, associated with active transcription are the trimethylation of histone H3 at lysine 4 (H3K4Me3) and the trimethylation of histone H3 at lysine 36 (H3K36Me3). Among the repressive marks are the trimethylation of histone H3 at lysine 27, 9, and 20 (H3K27Me3, H3K9Me2/3, and H3K20Me3). Increasing evidence suggests that alterations in the histone code may play an important role during prostate tumorigenesis and the expression levels of histone methyltransferases are often altered in multiple tumor types. In 2009, two independent groups have shown that, in CRPC models, enhanced levels of the active histone marks H3K4me1/2/3 are selectively enriched at the enhancer regions of some AR-regulated cell cycle genes thus promoting CRPC cell growth and survival [43, 44]. Further, an increase in the repressive histone marks H3K27me3 has also been shown during prostate cancer progression, correlating with a poor clinical outcome. H3K27me3 increase is attributed to the overexpression of the Enhancer of Zeste Homolog 2 (EZH2) and leads to the silencing of a wide number of genes (such as tumor suppressor genes, GAS2, PIK3CG, and ADRB2), whose repression can induce cancer cell growth and invasion. EZH2 is a histone methyltransferase belonging to the polycomb repressive complex 2 (PRC2) and its overexpression has been seen in metastatic prostate cancer [45] following microRNA-101 deletion, a negative regulator [46]. Despite several studies have focused on PCR2-mediated repression as EZH2 oncogenic function, in 2012, Xu et al. have demonstrated that EZH2 overexpression in a model of androgen-independent PCa (LNCaP-abl) correlates with a switch in EZH2 activity from gene repressor to gene activator. EZH2 was found to be highly phosphorylated on a specific serine residue (S21) and, depending on its phosphorylation, able to interact and act as an AR transcriptional co-activator. The importance of phosphorylation at S21 has also been confirmed for the androgen-independent growth of the androgen-sensitive cell line, LNCaP [47]. Moreover, H3K27me3 levels are significantly decreased in CRPC, further supporting the idea of an EZH2 oncogenic activity independently of its Polycomb repressive function [47]. A deregulated expression of EZH2 has also been described in other solid tumors such as: bladder, gastric, lung, and hepatocellular carcinoma. Interestingly, recent work showed that EZH2 can function as an upstream activator of another HMTase, MMSET/NSD2. NSD2 mediates H3K36me2, a histone mark associated with active transcription, and its expression and function has been reported to be tightly related to EZH2 in most human cancers [48]. In CRPC, NSD2 is overexpressed and acts as a strong co-activator of NF-κB, promoting cell proliferation and survival and tumor growth [49].
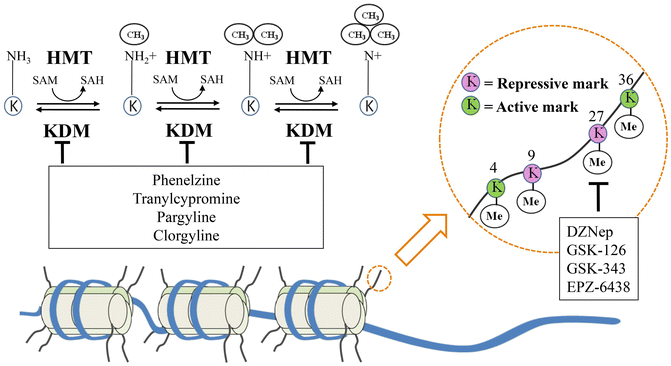
Fig. 20.3
Histone methylation: mechanism and functions of the principal methylation sites. Histones are the structural unit around which DNA is wrapped and packaged to form nucleosomes. Histone methylases (HMT) and demethylases (KDM) affects chromatin remodeling and function depending on the specific amino acid being modified and the extent of methylation. Active methylation marks lead to gene transcription while repressive marks induce gene silencing. Both EZH2 inhibitors (DZNep, GSK-126, GSK-343, and EPZ-6438) acting on the repressive mark on lysine 27 of histone 3, and KDMs inhibitors share promising therapeutic efficacy on the treatment of prostate cancer. Me Methyl group, HMT Histone methyltransferase, KDM Histone lysine demethylase, SAM S-adenosyl methionine, SAH S-adenosyl homocysteine, EZH2 Enhancer of zeste homolog 2
Histone Methyltransferases (HMTs) Inhibitors
Following those findings, small molecules targeting the histone methylation enzymes could have a significant impact on cancer treatment. Over the past years, several HMTs inhibitors have been developed and tested. Specifically, 3-Deazaneplanocin A (DZNep) is a cell-permeable compound, originally synthesized as S-adenosylhomocystein (AdoHcy) hydrolase inhibitor to indirect suppress SAM-dependent methylation, and has also been shown to deplete the levels of PRC2 components (EZH2, EED, and SUZ12). In cells of human acute myeloid leukemia (AML), DZNep treatment induced an increase of the cell cycle regulators p21, p27, and FBXO32, leading to cell cycle arrest and apoptosis. Moreover, a synergistic effect between DNZep and the HDAC inhibitor panobinostat as been demonstrated in human AML cells [50]. Among the other EZH2 inhibitors, GSK-126 and GSK-343 have been demonstrated to efficiently inhibit the growth of, respectively, diffuse large B-cell lymphoma (DLBCL) and epithelial ovarian cancer (EOC) cells. Since in hematological malignancies activating or inactivating mutations in EZH2 SET domain are often found, both inhibitors are effective against either wild type (WT) or mutant forms of EZH2. Another small molecule inhibitor (EPZ-6438) has been formulated for the treatment of non-Hodgkin lymphoma patients with oncogenic point mutation in EZH2. Based on the durable tumor regression seen in preclinical studies, this compound has been formulated to work as single-agent treatment and, in June 2013, a Phase 1/2 clinical trial of EPZ-6438 in patients with advanced solid tumors or with relapsed or refractory B-cell lymphoma has been initiated. In view of the promising results obtained in hematological disease, EZH2 inhibitors are now being tested also in prostate cancer.
Role of Histone Lysine Demethylases (KDMs) in CRPC
Histone lysine demethylases (KDMs) are enzymes able to remove both repressive and activating histone marks (Table 20.5). KDMs are grouped in seven major classes, each one targeting a specific histone methylation site. Belonging to the first class, KDM1, are two isoforms of flavin-dependent demethylases (KDM1A and B, also known as LSD1 and LSD2) responsible for the demethylation of H3K4me1/2. Since H3K4me2 is an active histone mark, KDM1 favors gene silencing. The second class of KDMs includes a cluster of six (KDM 2-7) of Fe2+/oxoglutarate-dependent enzymes, containing a characteristic Jumonji C (JmjC) domain. Most of the clusters include at least two members and are characterized by one or more targets.
Table 20.5
KDMs classes and functions
KDMs class | Target histone mark | Transcriptional modulation |
|---|---|---|
KDM1 | H3K4me1/2 | Silencing |
H3K9me2/3 | Activation | |
KDM2 | H3K36me2 | Silencing |
KDM3 | H3K9me2 | Activation |
KDM4 | H3K36me2/3 | Silencing |
H3K9me2/3 | Activation | |
KDM5 | H3K4me2 | Silencing |
KDM6 | H3K27me3 | Activation |
KDM7 | H3K9me1/2 and H3K27me1/2 | Activation |
Overexpression or mutation of KDMs has been linked to mis-erased histone methyl modifications and may play oncogenic or tumor-suppressive roles in several neoplasms. Histone demethylases such as KDM5A, 5C, and 6A are involved in several malignancies (acute myeloid leukemia, esophageal, renal, and squamous cell carcinomas) and KDM1A is thought to play a role in several neoplasms including PCa [51]. In PCa, KDM1A can function as AR co-activator and its oncogenic activity is partially due to its ability to trigger Myc-dependent transcription [51], and inhibit p53 pro-apoptotic function [52]. In the presence of high androgen concentrations, KDM1A can be recruited by the AR to mediate gene silencing thus acting as co-repressor [53]. In primary PCa, high KDM1A expression predicts higher risk of tumor relapse after prostatectomy [54]. Other KDMs observed to be overexpressed in CRPC are: KDM4C, found to be required for esophageal squamous carcinomas cell proliferation [55], PHF8 implicated in PCa metastatic disease [56] and KDM5B, an AR-co-activator [57]. Conversely, KDM2A is downregulated in PCa, indicating that it could play a tumor-suppressive function through its role in maintaining genome integrity (Table 20.6) [58]. Moreover, it has been shown that a correlation between KDMs expression and different PCa progression stages indicating that KDMs could have a role as novel biomarkers for prediction of tumor-initiation, progression-free survival, and androgen-independent state [59].
KDMs class | Expression in PCa | Role in PCa |
|---|---|---|
KDM1A | Overexpressed | AR co-activator/repressor |
KDM2A | Downregulated | Putative tumor suppressor |
KDM3A | Overexpressed | AR co-activator |
KDM4A | Overexpressed | |
KDM4B | Overexpressed | |
KDM4C | Overexpressed | AR co-activator |
KDM5B | Overexpressed | AR co-activator |
KDM5C | Overexpressed | TGFB signaling suppressor |
KDM6B | Overexpressed | Putative oncogene |
PHF8 | Overexpressed | Mediates cell invasion |
Histone Lysine Demethylases (KDMs) Inhibitors
KDMs can be targeted by selective small molecule inhibitors, which are already being tested in biochemical and preclinical models. Since KDM1A catalytic domain shares homology with neural Mono Amino Oxidase (MAO), pharmacological inhibitors developed as anti-depressive agents have been employed to target cancer cells. Indeed, phenelzine, tranylcypromine, and pargyline have been reported to act as KDM1A inhibitor. Pargyline has been first described as a KDM1A inhibitor in PCa cells, but further studies failed to confirm this observation. Tranylcypromine and its analogues (NCL-1 and NCL-2) have proved to more effectively inhibit H3K4me2 along with a safe toxicity profile. For this reason, tranylcypromine analogues may be particularly effective to prevent PCa recurrence and transition to an androgen-independent state [60]. Another MAO inhibitor, clorgyline, has shown to have anti-proliferative and pro-differentiation activity on epithelial cells derived from high grade-PCa and, more recently, the γ-pyronenamoline has been described to inhibit PCa proliferation both in vitro and in vivo via KDM1A suppression [61]. A small number of KDM4 inhibitors have been developed over the past last years, but none of them has been tested in PCa. In 2010, a series of hydroxamic acids targeting KDM4A/4C has been produced and tested. In vitro, these compounds demonstrated very low effect on PCa cells as single agents, but displayed synergistic activity in combination with the tranylcypromine analogue NCL-2 [62].
Noticeable, KDM1A and HDAC inhibitors have shown synergistic antitumor activity on glioblastoma cells [63]. The HDAC inhibitor vorinostat by inhibiting also EZH2 and H3K4 demethylases at micro-molar concentrations represents a promising epigenetic drug for cancer therapy.
miRNA and CRPC
MicroRNAs (miRNAs) are small non-coding RNA, which are approximately 19–22 nucleotides in length. In 1993 and 2000, the first two miRNAs, lin-4 and let-7, were identified in Caenorhabditis elegans [64, 65]. To date, there are 1,872 miRNAs that have been discovered in the human genome (miRBase: Released 20-June 2013). Furthermore, miRNAs have been shown to be involved in the regulation of cell differentiation, development, metabolism, cell cycle, and signaling transduction pathways by targeting 3′ untranslated region (3′ UTR) of target mRNA. This binding leads to degradation of target mRNAs. Dysregulation of miRNAs has been reported in infection diseases, cardiovascular diseases, neurodegenerative diseases, and cancer [66]. For this reason, numerous studies have investigated the function, underlying mechanisms, and therapeutic targeting of miRNAs in multiple disease types. Specific to CRPC, miRNAs have been demonstrated to be involved in disease progression, suggesting their utility as markers for diagnosis/prognosis, as well as novel therapeutic targets (Tables 20.7 and 20.8). MiRNA can also be divided into two categories, oncomir and tumor suppressor miRNA, according to their functions in promoting cell growth or inducing cell death.
Table 20.7
The in vitro studies of miroRNAs in prostate cancer
MicroRNA | Role in PCa | Cell lines | Targets | Functions | Reference |
|---|---|---|---|---|---|
miR-20a | Oncomir | PC3 | E2F1-3 | Apoptosis | [82] |
miR-21 | Oncomir | LNCaP; LAPC4 | PTEN; AKT; Androgen pathway | mTOR pathway; Androgen independency | [69] |
miR-27a | Oncomir | LNCaP | Prohibition | Androgen receptor pathway | [96] |
miR-32 | Oncomir | LNCaP | BTG2 | Apoptosis | [83] |
miR-106 | Oncomir | LNCaP | p21 | Cell cycle control | [97] |
miR-125 | Oncomir | PC3; DU145; PC-346C; LNCaP; LNCaP-cds2; LNCaP-R273H; | BAK1; PUMA; p53 | Apoptosis | [75] |
miR-141
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|