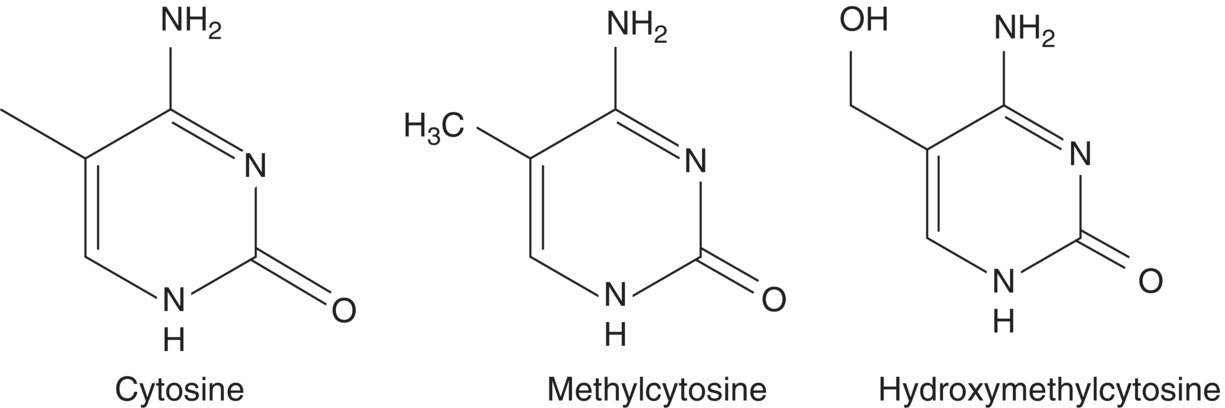
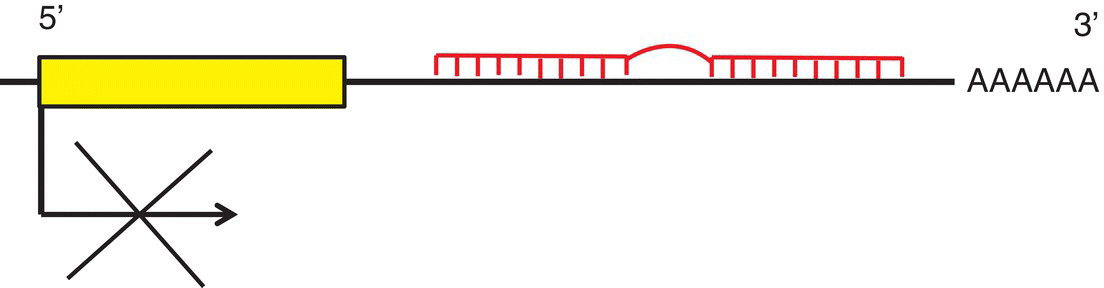
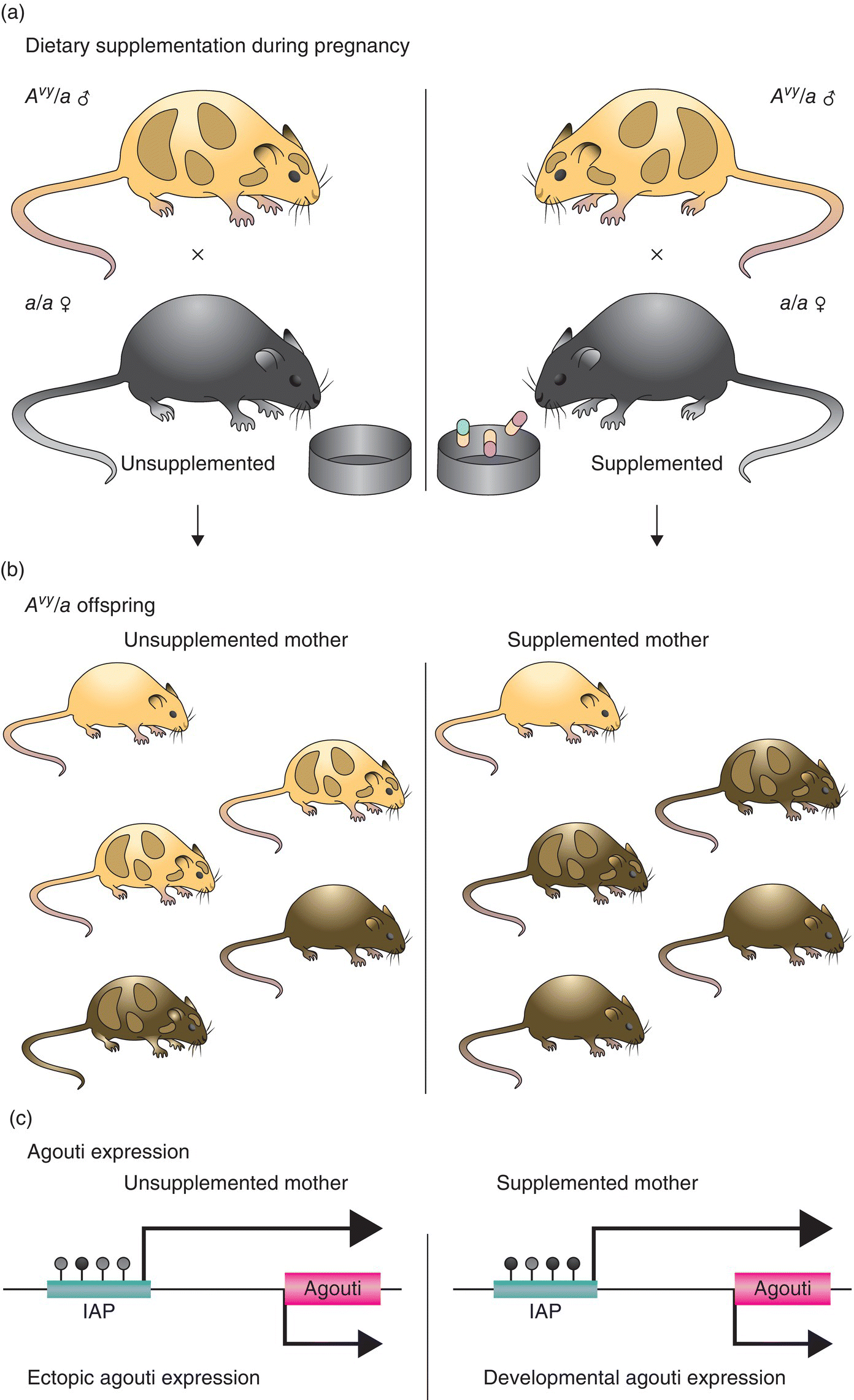
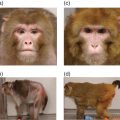
14 John C Mathers Newcastle University The human nuclear genome contains over 3 billion base pairs (bp) and the information to direct the synthesis of approximately 20 000 proteins. In addition, each mitochondrion within the cell contains about 16 600 bp, which encode just 37 genes. That said, any given human cell expresses only a small proportion of all the genes encoded in the genome. Most cells express a common set of genes, known as house-keeping genes, which are needed for generic purposes such as nutrient transport and adenosine triphosphate (ATP) generation. However, different cell types, such as neurones or hepatocytes, express consortia of genes that are characteristic of that cell type (described as cellular differentiation) and allow the cell to carry out the specific functions of the relevant cell type within particular tissues, for instance brain or liver. In addition, cells express distinctive complements of genes at different stages during development and in response to specific physiological states, for example epithelial cells in the breast during lactation. Cells also need to be able to respond appropriately to environmental factors such as dietary intake or exposure to chemical hazards. For all of these reasons, cells have evolved sophisticated, and dynamic, processes to ensure that the correct consortium of genes is expressed in a particular cell under the specific circumstances pertaining at that moment in time. Expression of genes can be regulated at any stage, from initial copying of the information within deoxyribonucleic acid (DNA) to make messenger ribonucleic acid or mRNA (transcription), through processing of ribonucleic acid (RNA) species and synthesis of the encoded proteins (translation) to post-translational processing of proteins. However, the key stages in the regulation of expression are regulating the amount of mRNA that is produced from a particular gene and then regulating the translation of that mRNA into protein. Such regulation operates over a very wide range of timescales. For example, some genes are expressed only during very early life (embryogenesis and fetal development) and then remain silent (switched off) during the rest of the life course. In contrast, expression of other genes is regulated over periods of minutes or hours, for instance genes in immune cells when exposed to an antigen and hepatocytes in response to feeding or fasting. Because of the criticality of ensuring that cells are equipped appropriately to carry out their function, it is not surprising that about 10% of all the proteins encoded in the human genome contain DNA-binding domains and are involved in regulating gene expression. Many of these proteins are transcription factors that, often in combination, provide relatively short-term regulation of gene expression. In addition, epigenetic processes exert further control over gene expression, which is essential for cell and tissue function and for health. In 1942, a decade before Crick and Watson discovered the structure of DNA and ushered in the era of molecular biology, C.H. Waddington coined the term ‘epigenetics’. Originally, epigenetics was used to describe how genes might interact with their environment to produce a particular phenotype. Nowadays, it is usually understood to be the study of the processes through which gene expression (and cellular phenotype) is regulated, that are heritable across cell generations and that do not involve changes in the DNA sequence. These processes involve a complex consortium of epigenetic marks and molecules that act in consort. The main epigenetic marks include chemical modifications of the cytosine base within DNA (Figure 14.1) and post-translational modifications of histones. The main epigenetic mark on DNA in animals is the addition of a methyl group to the 5′position on cytosine residues when that cytosine is followed by a guanine – that is, a so-called CpG dinucleotide – to produce 5′methylcytosine (5mC). In addition, it has been discovered very recently that methylated cytosines in CpG dinucleotides can be further modified by oxidation to form hydroxymethylcytosine (5hmC; Figure 14.1). Relatively high concentrations of 5hmC are seen in the brain and in embryonic stem cells. The role of 5hmC is not well understood, but it appears that it may be an intermediate in the demethylation of 5mC; that is, an important part of the dynamic process through which methyl marks can be added to, and removed from, DNA. Figure 14.1 Cytosine and derivatives, which are the principle epigenetic marks on DNA. Histones are the chief protein components of chromatin. Two of each of the core histones (H2A, H2B, H3 and H4) forms a globular protein complex around which 147 base pairs of DNA are wrapped in the nucleus, with a linker histone (H1) at each end locking the DNA in place. Protruding from the globular core are N-terminal protein ‘tails’ in which specific amino acid residues can be covalently modified by the addition of small chemical groups – that is, phosphate, methyl and acetyl groups – and by the addition of small regulatory proteins – that is, ubiquitin and SUMO (small ubiquitin-like modifier). These constitute the epigenetic ‘marks’ on histones. Epigenetic molecules fall into two groups. The first are the enzymes and other proteins that add or remove epigenetic marks and those that ‘read’ those marks to switch on, or switch off, expression of the corresponding gene. For example, this group of molecules includes DNA methyltransferase (DNMT) enzymes, which catalyse the addition of methyl groups to cytosines in DNA, and histone acetyl transferases (HATs) and histone deacetylases (HDACs), which catalyse the addition and removal, respectively, of acetyl groups on specific amino acids, for instance lysine, in histone tails. The second group of epigenetic molecules is composed of small RNAs (described as microRNA), which are typically 22 nucleotides long. These microRNA do not code for proteins, but regulate the expression of genes through base pairing with complementary sequences within the target mRNA species. In most cases, this binding to the 3′end of mRNA results in degradation of the target mRNA and, because this means that no protein is synthesised, the corresponding gene is effectively silenced (Figure 14.2). In mammals, there is only partial complementarity between the miRNA sequence and that of the corresponding mRNA. As a consequence, a given microRNA may have multiple mRNA targets and translation of a given mRNA may be regulated by a combination of microRNAs. Figure 14.2 Binding of a microRNA molecule to a complementary sequence within a target mRNA species, causing gene silencing. The microRNA is shown in the centre. The box on the left represents the sequence within the mRNA that encodes the protein and the black cross indicates that binding of the microRNA to the 3′end of the mRNA transcript prevents translation of that protein. Epigenomics is the study of the totality of epigenetic marks and molecules within a cell or tissue, which is described as the epigenome. This is analogous to the terms genomics, transcriptomics, proteomics and metabolomics, which describe all the genes, mRNA species, proteins and metabolites, respectively, in a cell or other biological sample and are discussed in more detail in Chapter 13. Following the successful projects to map the human genome and to identify the genetic variants, such as single nucleotide polymorphisms (SNP) and copy number variants (CNV), in the human genome, there are now consortia attempting to map specific epigenomes. For example, the Human Epigenome Project (HEP) aims to identify, catalogue and interpret genome-wide DNA methylation patterns of all human genes in all major tissues (for details, see http://www.epigenome.org/). The genome of the fertilised human egg contains all the genetic information to direct development of the human adult, who contains over 200 different cell types, all of which contain exactly the same DNA but carry out distinctively different functions. Asymmetrical cell division leading to different cell types (cellular differentiation) occurs during embryogenesis and is very carefully orchestrated to ensure that each cell type expresses its characteristic complement of genes. This is achieved via chromatin, the DNA–nucleosome polymer, which is a dynamic molecule capable of existing in many configurations. The combination of epigenetic marks and molecules (epigenome) determines the higher-order organisation of chromatin. Those chromatin domains that are highly compacted (known as heterochromatin) contain genes that are silenced, whereas the more open chromatin regions (known as euchromatin) allow access by the transcriptional machinery and gene expression. In the same way that optimum health requires each of the body’s organs and tissues to do its particular job efficiently in response to whatever challenge the body faces, so, at the cellular level, the proper function of each individual cell requires expression of the consortium of genes that is appropriate for that cell type, for instance an endothelial cell or an enterocyte, in its given environment. In addition, cells need to be able to respond correctly to changing conditions. For example, if we consume potentially hazardous chemicals (xenobiotics), hepatocytes may need to upregulate the expression of defence genes, such as those of the cytochrome P450 superfamily, to enable the body to detoxify those chemicals. Similarly, cells can save energy and other resources by shutting down the expression of genes that are not needed at that particular time. In summary, our genotype is fixed, but our phenotype is plastic and responds to life’s experiences and exposures. It is now established that several dietary factors, and other environmental exposures, result in changes in epigenetic marks and molecules, some of which lead to alterations in gene expression (see Table 14.1). Table 14.1 Examples of dietary factors and nutritional status that influence epigenetic marks (see McKay and Mathers 2011 for a detailed review). In many cases, the strongest evidence for the impact of nutrition on epigenetic regulation of gene expression comes from studies using model systems (mammalian cells and animal models), but there is growing evidence for similar effects from studies carried out in humans. This has led to the hypothesis that epigenetic processes are potentially important mechanisms through which dietary exposures and nutritional status may influence health. In particular, this is an attractive hypothesis for explaining how nutrition can have very long-term effects on health. Because at least some epigenetic marks such as patterns of DNA methylation are copied from one cell generation to the next, altered DNA methylation caused by early-life nutrition could result in lifelong changes in gene expression, altered risk of disease and effects on life span. This is best illustrated by studies of the Agouti mouse. The coat colour of the offspring in this model is influenced by the supply of folate and other methyl donors provided to the dam during pregnancy (see Figure 14.3). Supplemented dams produce a higher proportion of offspring with darker (brown) coats rather than the predominantly yellow-coloured offspring of unsupplemented dams. This change in coat colour is due to altered expression of the Agouti gene caused by methylation of a regulatory region upstream of the gene. These effects of the maternal diet on DNA methylation are seen in every tissue of the body and the effects on coat colour endure throughout the animal’s life. In this mouse model, maternal methyl donor supply affects not only coat colour but also other important phenotypical characteristics. For example, the ‘yellow’ mice tend to be fatter and more susceptible to obesity-related diseases such as type 2 diabetes than their brown litter mates. Interestingly, maternal exposure to genistein (an isoflavone from soybeans) and to bisphenol A (an industrial chemical used in production of polycarbonate plastics) results in similar changes in DNA methylation and in coat colour in Agouti mice, although neither genistein nor bisphenol A is a methyl donor. Figure 14.3 Maternal supplementation of Agouti mice with methyl donors (folate, betaine, vitamin B12 and choline ; Panel (a)) results in litters with a higher proportion of offspring with dark (brown) coats, whereas the offspring of the unsupplemented dams have predominantly yellow coats (Panel (b)). Panel (c) shows the molecular mechanism for this nutritionally induced phenotypical change. The pale grey bar represent the regulatory region (labelled IAP) upstream of the Agouti gene (shown on the right). Maternal supplementation results in greater methylation of DNA in the regulatory region, shown as black-filled circles above the IAP region. Reprinted by permission from Macmillan Publishers Ltd: Nat Rev Genet. Jirtle RL & Skinner MK. Environmental epigenomics and disease susceptibility, 8(4):253-62, copyright 2007. These ideas are conceptualised in the 4Rs of nutritional epigenomics model (Mathers 2008), illustrated in Figure 14.4. In this model, exposure to specific dietary factors produces marks on the genome such as an altered pattern of DNA methylation; that is, the ‘signal’ is Received and Recorded. To have long-term effects, there needs to be a mechanism through which evidence of that exposure is Remembered when the cell divides. In the case of DNA methylation, this memory mechanism is the copying of patterns of DNA methylation from the parental strand of DNA to the daughter strand by the enzyme DNMT1, which occurs immediately after synthesis of the new (daughter) DNA strand during mitosis. Since at least some changes in DNA methylation (and other epigenetic marks) result in changes in gene expression, this is the mechanism through which the functional consequences of the original nutritional exposure are Revealed (Figure 14.4
Epigenetics
14.1 Introduction: The human genome and the regulation of gene expression
14.2 What is epigenetics?
14.3 Epigenetic marks and molecules
14.4 Epigenetic regulation of gene expression
14.5 Nutrition and epigenetics
Dietary factor
Epigenetic change
Folate and other methyl donors
Multiple effects on methylation of specific genes and on methylation and acetylation of histones
Butyrate
Extra butyrate increased acetylation of histones H3 and H4
Isoflavones
Isoflavone supplementation induced dose-dependent changes in methylation of genes in the mammary gland
Protein
Low protein intake during pregnancy caused reduced methylation of the promoters of several genes, including the glucocorticoid receptor (GR) and the peroxisomal proliferator-activated receptor alpha (PPARα) genes
Selenium
Supplemental selenium increased DNA methylation in the liver
Polyphenols
Polyphenols may alter patterns of DNA methylation by affecting expression of key enzymes, including DNMT
Obesity
Patterns of DNA methylation appear to be influenced by obesity and to predict responders to weight-loss interventions
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel


Full access? Get Clinical Tree


