Epigenetic mechanisms, such as DNA methylation and histone modifications, drive stable, clonally propagated changes in gene expression and can therefore serve as molecular mediators of pathway dysfunction in neoplasia. Myelodysplastic syndrome (MDS) is characterized by frequent epigenetic abnormalities, including the hypermethylation of genes that control proliferation, adhesion, and other characteristic features of this leukemia. Aberrant DNA hypermethylation is associated with a poor prognosis in MDS that can be accounted for by more rapid progression to acute myeloid leukemia. In turn, treatment with drugs that modify epigenetic pathways (DNA methylation and histone deacetylation inhibitors) induces durable remissions and prolongs life in MDS, offering some hope and direction in the future management of this deadly disease.
The myelodysplastic syndromes (MDSs) are a group of diverse and heterogeneous syndromes characterized by clonal proliferation, bone marrow failure, and an increased risk of development of acute myelogenous leukemia (AML). The natural course of the disease ranges from slow progression to rapid evolution into AML. There are several classification systems for MDS, such as the International Prognostic Scoring System, the World Health Organization classification, and more recent clinical schemes, but none captures the full heterogeneity of the disease. Heterogeneity and progression are likely related to molecular changes that drive the disease phenotype and biology, but these remain incompletely understood. Insights into the pathogenesis of cancer has come from studying genetic changes in neoplastic cells. In MDS, several cytogenetic abnormalities have been identified that characterize subgroups of patients, such as deletions of chromosome 5 or 7 in patients with poor prognosis, isolated deletion of 5q, or trisomy 8. Mutations of several genes also characterize subsets of cases, including RAS, TET2, RUNX1, and other genes. Despite these advances, the molecular causes of MDS and its peculiar clinical features remain poorly understood. Epigenetic changes have been recognized in the past decade as major drivers of the malignant phenotype. Epigenetics refers to the study of clonally inherited changes in gene expression without accompanying genetic changes. There are 3 general molecular mechanisms carrying epigenetic information—DNA methylation, histone modifications, and RNA interference. In cancer, there is particular recent interest in the involvement of aberrant DNA methylation and histone modifications in gene silencing that then mediate altered physiology. They may be particularly relevant to MDS pathogenesis given that the disease responds well to drugs that affect DNA methylation, a major epigenetic modulator. In this article, recent progress in MDS epigenetics and epigenetic-based therapies is reviewed.
DNA methylation in epigenetic regulation
DNA methylation is a covalent modification of cytosines resulting in the formation of 5-methylcytosine (5mC), a base that changes the interactions between protein and DNA. In adult mammalian cells, DNA methylation normally affects cytosine when it is part of the cytosine-phospho-guanine (CpG) dinucleotide. CpG methylation can occur anywhere in the genome but is particularly relevant when it involves CpG rich regions called CpG islands. In turn, these can be present in about half of human gene promoters. CpG island methylation is associated with absent transcription from the involved promoter, and this silencing is stably transmitted through mitosis, thus ensuring clonal inheritance. This association between CpG island methylation and absent transcription is most striking when considering the 2 physiologic conditions where this process was described, X-chromosome inactivation and imprinting. In both cases, 1 of the 2 copies of the involved genes is transcriptionally silent in association with promoter methylation, despite continued normal expression of the unmethylated allele. Evidence for a direct role of methylation in maintaining the silenced state is from studies where methylation was relieved via pharmacologic or genetic reduction in DNA methyltransferase (DNMT) activity. In most such studies, biallelic expression could be restored, in association with decreased methylation of the affected promoters.
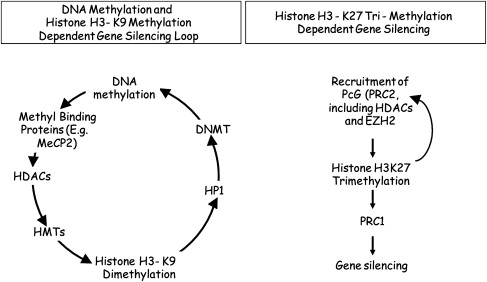
The mechanism whereby CpG island methylation suppresses gene transcription recently was partially elucidated ( Fig. 1 ), at least in vitro. Methylated CpG islands form excellent binding sites for methylated DNA-binding proteins (often with transcriptional repression properties), such as MeCp2. MeCp2 binding is followed by the recruitment of a protein complex that includes histone deacetylases (HDACs) and eventually leads to a closed chromatin configuration. This closed chromatin configuration results in exclusion of transcription factors, thus ensuring allele-specific inactivation. Methylation-related epigenetic silencing also is associated with histone H3 lysine 9 (H3K9) methylation. Evidence suggests that H3K9 methylation is a critical modification that is associated with closed chromatin at DNA methylation sites, and it was proposed that a cascade of events follows DNA methylation (MeCP2 binding, H3K9 deacetylation, and H3K9 methylation) and ensures transcriptional suppression (see Fig. 1 ). Separately, DNMT1 can directly suppress transcription (without DNA methylation) through interactions with HDACs. H3K9 methylation itself seems to set up a silencing loop by attracting more DNA methylation and may sometimes precede hypermethylation.
DNA methylation changes in cancer
There are complex changes in DNA methylation in cancer. For the most part, these changes involve simultaneous global demethylation, increased expression of DNMTs, and de novo methylation at previously unmethylated CpG islands. Demethylation was first discovered by studying overall 5mC content in tumors and seems to involve primarily satellite DNA, repetitive sequences, and CpG sites located in introns. The cause of this demethylation remains unclear, although it could be related to alterations in proliferation or cell-cycle control. The functional consequences of hypomethylation are not entirely clear, but there is mounting evidence that gene-specific hypomethylation can cause increased expression of various genes that could contribute to the neoplastic phenotype. An increased mutation rate was demonstrated in cells in which severe hypomethylation (>75%) was achieved by homozygous deletion of DNMT1, but it is not clear whether or not this degree of hypomethylation is ever achieved in neoplasms.
Increased enzymatic DNMT activity is a property of nearly all transformed cells. Increased mRNA levels for DNMT1, DNMT3a, and DNMT3b also are described in some neoplasms, including leukemias, and these 3 DNMT genes probably account for the observed increase in activity. The causes and functional significance of these increases remain unclear. DNMTs seem to be cell-cycle regulated, and it is argued that DNMT levels reflect the physiologic state of increased proliferation in neoplasia. Alternatively, DNMT1 is reported to increase after oncogene activation, and it is possible that its levels in neoplasia reflect the various molecular defects seen in tumors. The functional significance of increased DNMT activity is also poorly defined. In several systems, increased DNMT activity has been found to be transforming, but it is not clear whether or not this is due to increased CpG island methylation and tumor-suppressor gene silencing or to direct effects on the cell cycle. The facts that in primary tumors and cell lines no correlation was found between DNMT activity and gene silencing and that DNMT-related transformation is reversible when the oncogenic stimulus is removed support the latter possibility. Nevertheless, simultaneous inhibition of several DNMTs does inhibit cancer cell growth, making them potential therapeutic targets.
In parallel to global hypomethylation and increased DNMT activity, there also are distinct and frequent localized increases in methylation, often involving CpG islands. Because CpG island methylation is associated with repressed transcription that is stably inherited through mitosis, this de novo methylation in transformed cells is proposed as an alternate mechanism for inactivating tumor-suppressor genes. Several genes are shown to be transcriptionally silent in neoplasia in association with CpG island methylation, including in leukemias. The most convincing evidence for CpG island methylation as a true alternative to mutations in neoplasia is from studies of RB1 , p16 , VHL , and MLH1 . For each of these genes, the tumor-spectrum of methylation events is virtually the same as that for mutations, and there are described cases where 1 allele of the gene is inactivated by methylation whereas the other is mutated, suggesting an equivalent growth advantage for each event in neoplasia. For example, in colorectal cancer, the HCT116 cell line carries 1 mutated unmethylated p16 allele whereas the second allele is unmutated but densely hypermethylated and transcriptionally silent. Moreover, the MLH1 mismatch repair gene is unmutated but densely methylated in the mismatch repair deficient cell line RKO, and nearly normal levels of mismatch repair can be restored by inhibiting DNMT activity.
DNA methylation changes in cancer
There are complex changes in DNA methylation in cancer. For the most part, these changes involve simultaneous global demethylation, increased expression of DNMTs, and de novo methylation at previously unmethylated CpG islands. Demethylation was first discovered by studying overall 5mC content in tumors and seems to involve primarily satellite DNA, repetitive sequences, and CpG sites located in introns. The cause of this demethylation remains unclear, although it could be related to alterations in proliferation or cell-cycle control. The functional consequences of hypomethylation are not entirely clear, but there is mounting evidence that gene-specific hypomethylation can cause increased expression of various genes that could contribute to the neoplastic phenotype. An increased mutation rate was demonstrated in cells in which severe hypomethylation (>75%) was achieved by homozygous deletion of DNMT1, but it is not clear whether or not this degree of hypomethylation is ever achieved in neoplasms.
Increased enzymatic DNMT activity is a property of nearly all transformed cells. Increased mRNA levels for DNMT1, DNMT3a, and DNMT3b also are described in some neoplasms, including leukemias, and these 3 DNMT genes probably account for the observed increase in activity. The causes and functional significance of these increases remain unclear. DNMTs seem to be cell-cycle regulated, and it is argued that DNMT levels reflect the physiologic state of increased proliferation in neoplasia. Alternatively, DNMT1 is reported to increase after oncogene activation, and it is possible that its levels in neoplasia reflect the various molecular defects seen in tumors. The functional significance of increased DNMT activity is also poorly defined. In several systems, increased DNMT activity has been found to be transforming, but it is not clear whether or not this is due to increased CpG island methylation and tumor-suppressor gene silencing or to direct effects on the cell cycle. The facts that in primary tumors and cell lines no correlation was found between DNMT activity and gene silencing and that DNMT-related transformation is reversible when the oncogenic stimulus is removed support the latter possibility. Nevertheless, simultaneous inhibition of several DNMTs does inhibit cancer cell growth, making them potential therapeutic targets.
In parallel to global hypomethylation and increased DNMT activity, there also are distinct and frequent localized increases in methylation, often involving CpG islands. Because CpG island methylation is associated with repressed transcription that is stably inherited through mitosis, this de novo methylation in transformed cells is proposed as an alternate mechanism for inactivating tumor-suppressor genes. Several genes are shown to be transcriptionally silent in neoplasia in association with CpG island methylation, including in leukemias. The most convincing evidence for CpG island methylation as a true alternative to mutations in neoplasia is from studies of RB1 , p16 , VHL , and MLH1 . For each of these genes, the tumor-spectrum of methylation events is virtually the same as that for mutations, and there are described cases where 1 allele of the gene is inactivated by methylation whereas the other is mutated, suggesting an equivalent growth advantage for each event in neoplasia. For example, in colorectal cancer, the HCT116 cell line carries 1 mutated unmethylated p16 allele whereas the second allele is unmutated but densely hypermethylated and transcriptionally silent. Moreover, the MLH1 mismatch repair gene is unmutated but densely methylated in the mismatch repair deficient cell line RKO, and nearly normal levels of mismatch repair can be restored by inhibiting DNMT activity.
Histone modifications and the histone code
Histones are small proteins that form a core around which DNA is wrapped, forming nucleosomes. Nucleosomes are the basic in vivo structural unit of DNA and consist of 8 histone molecules (2 each of histones H2A, H2B, H3, and H4) around which a loop of DNA is wrapped. Although H2A and H2B are thought to play primarily a structural role, it has become apparent that histones H3 and H4 are key integrators of a variety of signals that regulate gene transcription. In particular, these 2 histone proteins have histone tails, or strings of amino acids, that protrude outside the basic nucleosomal structure and make contact with DNA. Specific post-translational modifications of the amino acids in these histone tails (eg, methylation, acetylation, phosphorylation, ubiquitination, and sumoylation) interact with other proteins to create nucleosomes that are relaxed and promote transcription or nucleosomes that are closed, exclude transcription factors, and result in gene silencing. These histone modifications occur dynamically and are mediated by specific histone-modifying proteins. Targeting of these histone modifiers to specific gene promoters in turn is achieved by transcriptional activator/coactivator complexes in response to physiologic stimuli. Thus, histone modifications form a code that integrates gene activation/inactivation/silencing signals, such that the transcriptional activity of a given promoter can be predicted by looking at the specific histone modifications.
Currently, the best understood histone modifications are acetylation and methylation of specific residues, although it is clear that other modifications also play a role in the process. Acetylation of specific residues on histones H3 and H4 is typically associated with active gene transcription. Methylation of specific residues can be associated with activation of transcription (H3 lysine 4 [H3K4]) or silencing (H3K9 and H3K27). The different modifications also significantly interact. Thus, H3K4 methylation promotes H3K9 acetylation. Moreover, H3K9 can be acetylated or methylated (but not both), and the switch from acetylation to methylation at this residue seems to be a key element of silencing across many organisms. H3K9 promotes silencing by recruiting HP1 and a silencing complex that changes chromatin structure locally and results in exclusion of transcription factors. An added complexity to the process was demonstrated when residues were shown to have several possible methylation states. For H3K9, mono- and dimethylation are associated with euchromatin silencing, and trimethylation is associated with pericentromeric heterochromatin silencing. The H3K9 methylation/silencing switch seems conserved across evolution and is active in mammalian cells. H3K27 methylation seems distinct from H3K9 methylation; H3K27 trimethylation is reported to occur early in X-chromosome inactivation and leads to silencing by recruitment of polycomb group (PCG) proteins. In contrast, H3K9 trimethylation is primarily a mark of heterochromatin and leads to silencing by recruitment of HP1 proteins. Thus, these 2 processes, which are mediated by different enzymes, are potentially functionally distinct. There is considerable interest in the involvement of the histone code in stemness and differentiation. A bivalent chromatin domain is described in embryonic stem cells, whereby some unexpressed genes have coexistence of activating (H3K4Me) and silencing marks (H3K27Me). On differentiation, this bivalency resolves into the activated or the suppressed state.
Although histone modifications are stable, histones are eventually degraded, and the nucleosome structure is substantially altered during DNA synthesis. Moreover, histone acetylation and methylation can be directly reversed by many HDACs and histone demethylases. Thus, for histone modifications to retain epigenetic memory, some form of targeting has to be operative. DNA methylation is one such form of targeting as is persistent expression of transcription factors. It also is argued that the histone modifications themselves could be a form of epigenetic memory in the absence of DNA methylation or other proteins to direct the process.
Histone modifications and cancer
Histone modifications are implicated in the neoplastic process in a variety of ways. Indirectly, several genes that are altered in cancer affect gene expression via recruitment of histone-modifying enzymes and resulting changes in gene expression. For example, the PML-RARalpha gene translocation in acute promyelocytic leukemia recruits HDACs to inhibit the expression of target genes, thus contributing to malignant transformation. More directly, several histone-modifying enzymes are molecularly altered in cancer. For example, the histone acetyltransferase CBP is mutated in some acute leukemias ; the MLL gene, an H3K4 methylase, is rearranged in a significant portion of acute leukemias ; the RIZ1 gene, a putative histone methylase, is silenced in some cancers ; and EZH2, an H3K27 trimethylase, is overexpressed in various malignancies, which also exhibit de novo H3K27triM mediated silencing. Another mechanism modifying the histone code in cancer is aberrant promoter methylation. Such DNA methylation may lead to silencing via recruitment of histone-modifying enzymes that ultimately alter the histone code in favor of gene silencing.
Epigenetic changes in MDS
Most studies of epigenetics in MDS thus far have focused on DNA methylation. Several genes are transcriptionally silenced in association with promoter DNA methylation in this disease ( Table 1 ). These include genes involved in cell-cycle regulation (CDKN2A), apoptosis (DAPK1 and RIL), adhesion and motility (CDH1 and CDH13) and other pathways. Some of these genes clearly have minimal functional impact on the disease, because they are not expressed in normal hematopoietic cells. MDS cases often show hypermethylation of several genes simultaneously. Thus, hypermethylation can be viewed in a similar way as mismatch repair deficiency and microsatellite instability in cancer: many loci are affected simultaneously, a few of which likely have functional consequences.
| Gene | Methylation Frequency (%) | Function | Note |
|---|---|---|---|
| Calcitonin | 50 | Differentiation | |
| CDKN2B | 23–80 | Cyclin-dependent kinase inhibitor; cell cycle/proliferation | Tumor-suppressor; methylation correlates with poor prognosis and progression to AML |
| DAPK | 50 | Proapoptotic serine/threonine kinase | |
| RASSF1 | 9 | Negative regulator of RAS signaling | Tumor-suppressor |
| FHIT | 50 | Purine metabolism | Putative tumor-suppressor; methylation correlates with poor prognosis and progression to AML |
| HIC1 | 32 | Transcriptional repressor | Tumor-suppressor |
| CDH1 | 15–27 | Adhesion and motility | Methylation correlates with poor prognosis and progression to AML |
| CTNNA1 | 10 | Alpha catenin | |
| ERα | 7–19 | Estrogen receptor | Methylation as part of a panel of genes (also including CDH1 and CDKN2A) correlates with poor prognosis and progression to AML |
| RIL | 36–70 | Proapoptotic, tumor suppressor | |
| CDH13 | 21 | Adhesion and motility | |
| NOR1 | 15 | Oxidored-nitro domain-containing protein | |
| NPM2 | 20 | Nucleophosmin/nucleoplasmin 2, involved in development | |
| OLIG2 | 41 | Basic helix-loop-helix transcription factor | |
| PGRA | 45 | Progesterone receptor | |
| PGRB | 45 | Progesterone receptor |
In MDS, CDKN2B (P15) is the most extensively studied gene. CDKN2B was reported methylated in 30% to 80% of the cases, with the variability likely due to different methods of measurement as well as inclusion of different types of MDS. Thus, CDKN2B methylation is reported as frequent in therapy-related MDS and in chronic myelomonocytic leukemia , refractory anemia with excess blasts in transformation, or AML arising from MDS. CDKN2B methylation in MDS also is associated with older age, deletions of 5q and 7q, and a poor prognosis. When present, CDKN2B methylation in MDS affects several lineages from clonogenic cells to circulating mononuclear cells. In a mouse model, loss of CDKN2B was associated with enhanced myeloid progenitor and reduced erythroid progenitor formation, suggesting that its inactivation plays a functional role in MDS.
In a recent study focusing on quantitative analysis of the methylation status of 10 separate genes, a hypermethylator phenotype was identified that marks a subset of cases with MDS. This phenomenon, first described in colon cancer, results in the simultaneous inactivation of several genes by an unknown mechanism. In MDS, this form of intense hypermethylation is associated with rapid progression to AML and a shortened survival in multivariate analyses. This explains in part why the methylation of so many genes is reported as prognostic in MDS (see Table 1 ): in all likelihood, all these studies of individual genes are pointing to a common subset of cases affected by the hypermethylator phenotype.
Whole genome scans for DNA methylation are beginning to reveal the complexity of the disease at an epigenetic level. Hundreds of genes are frequently hypermethylated in MDS, with evidence for enrichment of WNT pathway genes. As indicated by studies of individual genes, hypermethylation across the genome is associated with poor prognostic features and transformation to AML. There was a distinct methylation pattern in MDS and related AMLs compared with de novo AML, pointing to distinct pathogenic mechanisms. Thus, a consensus has emerged that DNA methylation is abnormal early on in MDS and that progression of the disease is associated with accumulation of additional epigenetic events.
In contrast to DNA methylation, detailed studies of histone modifications in MDS remain to be described. A few cases of AML have chromosomal translocations involving MLL1, a known epigenetic modifier. Recently, mutations in the polycomb-related gene ASXL1 were described in about 10% of cases, and mutations in the TET2 gene were found in about 25% of cases. TET2 is related to TET1, a gene that converts 5mC to 5-hydroxymethylcytosine, and thus may be involved in regulation of DNA methylation. It is not known whether or not MDS cases with abnormalities in MLL1, ASXL1, or TET2 have characteristic epigenetic patterns.
Related posts:
 Mouse Models of Myelodysplastic Syndromes
Mouse Models of Myelodysplastic Syndromes
 Innate Immune Signaling in the Myelodysplasti c Syndromes
Practical Recommendations for Hypomethylating Agent Therapy of Patients With Myelodysplastic Syndromes
Innate Immune Signaling in the Myelodysplasti c Syndromes
Practical Recommendations for Hypomethylating Agent Therapy of Patients With Myelodysplastic Syndromes
 Prognostic Classification and Risk Assessment in Myelodysplastic Syndromes
Prognostic Classification and Risk Assessment in Myelodysplastic Syndromes
 Myelodysplastic Syndromes Classification and Risk Stratification
Myelodysplastic Syndromes Classification and Risk Stratification
 Patient Selection for Transplantation in the Myelodysp lastic Syndromes
Patient Selection for Transplantation in the Myelodysp lastic Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


