Continents
Incidence
Mortality
North America
Male
15.5
3.7
Female
8.3
1.6
Both
11.7
2.6
Europe
Male
12.3
4.8
Female
5.9
1.9
Both
8.8
3.1
Oceania
Male
11.0
2.8
Female
5.3
1.3
Both
8.0
2.0
Latin America & Caribbean
Male
4.7
2.5
Female
2.5
1.2
Both
3.5
1.8
South America
Male
5.1
2.8
Female
2.7
1.3
Both
3.8
2.0
Asia
Male
3.8
1.7
Female
1.9
0.9
Both
2.8
1.3
In the United States, kidney cancer is the sixth most common cancer in men and eighth most common in women, with an estimated 63,920 new cases and 13,860 deaths in 2014 [3]. The age-adjusted incidence rate in men (21.0 per 100,000 person-years) is nearly twice as high as in women (10.6 per 100,000 person-years). Likewise, the mortality rate of men (5.8 per 100,000 person-years) is double that of women (2.6 per 100,000 person-years) [4]. During the past three decades, the incidence rate of kidney cancer has been steadily increasing at over 2 % per year (Fig. 1.1). The rise in incidence has been more rapid in blacks than in whites, especially in males. In contrast, mortality rates have been indiscriminate among blacks and whites since the early 1990s [5]. This raises the possibility that early-stage tumors with improved prognosis account for the excess in kidney cancer incidence among blacks. The etiology of RCC differs from that of other kidney cancers, and RCC is the major histologic type; thus, we only focus on epidemiology of RCC for the remainder of this chapter, and kidney cancer and RCC are used interchangeably.
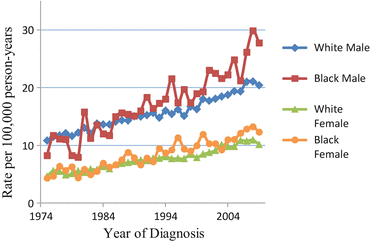
Fig. 1.1
Trends in age-adjusted incidence of kidney cancer by race and sex, 1974–2009 (Based on nine areas: San Francisco, Connecticut, Detroit, Hawaii, Iowa, New Mexico, Seattle, Utah, and Atlanta [3]. The rate is age-adjusted to the 2000 US Standard Population)
Several modifiable risk factors have been established for RCC, including obesity, cigarette smoking, and hypertension [1]. There is a rapidly growing body of evidence showing that energy balance may play a critical role in the development of the disease. However, due to inconsistent results reported by different studies, more solid evidence is needed to establish the association of RCC risk with energy intake, diet/nutrients, physical inactivity, and weight change. Other potential modifiable risk factors for which association is inconclusive include alcohol consumption, type II diabetes, occupational or environmental exposures, etc.
Systematic study of genetic variants is promising with the advent of genome-wide association study (GWAS) and next-generation sequencing (NGS). A few RCC susceptibility loci have been identified through recent GWASs which focus on common variants (minor allele frequency >0.05). Currently, four susceptibility loci have been confirmed by large global consortiums. Although GWAS has been successful in identifying susceptibility loci that are replicable, the total heritability explained by these loci is small. Additionally, a large proportion of single nucleotide polymorphisms (SNPs) are located in noncoding areas. The functions of tagging SNPs or SNPs in high linkage disequilibrium in these regions are not fully understood. On the other hand, thanks to the advent of next-generation sequencing and lowering costs, we are now able to identify variants with low and rare frequency (minor allele frequency of 0.1–1 % and <0.1 %, respectively) but larger effect size. Furthermore, intermediate phenotypic biomarkers are also being evaluated for their utility in predicting the predisposition of the disease. A number of intermediate phenotypic biomarkers in peripheral blood leukocytes (PBLs) have been linked to RCC risk, such as mitochondrial copy number change, telomere length, and suboptimal DNA repair capacity. Less is known about the etiologic role played by gene–gene and gene–environment interactions that could explain some extent of missing heritability. In this chapter, we review the current state of knowledge on the epidemiology of RCC in detail.
1.2 Modifiable Risk Factors
1.2.1 Cigarette Smoking
Cigarette smoking has been recognized as a causal risk factor with moderate effect on RCC. As early in the 1960s, one hospital-based case-control study has linked tobacco use to RCC risk for both genders [6]. The association was significant in all types of tobacco users. Since then, various studies have confirmed the association in both case-control and prospective cohort studies. A large meta-analysis of 5 cohort and 19 case-control studies reported a 50 % increase in RCC risk among male smokers and a 20 % increase among female smokers. A dose–response relationship between the number of cigarettes smoked and RCC risk was reported for both genders. Smoking cessation should be promoted because an approximate 15–30 % reduced risk was observed in both men and women who stopped smoking for more than 10 years [7]. The attributable risk percentage (AR%) of smoking calculated from population-based case-control and cohort studies is comparable (21 vs. 23 %), which indicates that more than one fifth of RCC could be prevented in the general population if they had no exposure to smoking. Although smoking is primarily a lifestyle factor in ever smokers, it is an environmental exposure to never smokers. One study showed that patients who were never smokers were more likely to report exposure to environmental tobacco smoking (ETS) either at home or in public [8]. However, interpretation of these results requires caution due to the likelihood of recall bias and the relatively small sample size.
A number of potential mechanisms linking smoking to the development of RCC have been proposed [1]. Cigarette smoke contains more than 60 carcinogens and most of them are formed during combustion [9]. One crucial mechanism of linking tobacco carcinogens with cancer is through the formation of DNA adducts. Benzo[α]pyrene (BaP), a polycyclic aromatic hydrocarbon (PAH), was the first carcinogen found in cigarette smoke, and its carcinogenicity has been confirmed in animals and humans. However, the level of BaP is much lower in cigarette products nowadays, and its carcinogenicity is weaker compared to some other PAH compounds [10]. Considerable evidence has linked cigarette-smoke nitrosamines such as 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), N′-nitrosonornicotine (NNN), and aromatic amines such as 4-aminobiphenyl to several cancer types. However, little supporting evidence has been gained from studies of kidney cancer. Only two studies have been conducted, and these data suggest that peripheral lymphocytes derived from patients with RCC are more sensitive to NNK or benzo[α]pyrene diolepoxide (BPDE) than those derived from healthy controls [11, 12]. In addition, smoking-induced chronic hypoxia of renal tissue caused by carbon monoxide exposure, tubulotoxicity, increased oxidative stress, and endothelial cell dysfunction are all hypothesized to increase RCC risk [1].
Due to the great efforts in the campaign to target smoking cessation and changes of policy in public health, the overall prevalence of cigarette smoking has declined over the past several decades in the United States. By contrast, incidence rates of kidney cancer have steadily increased at more than 2 % per year since the 1970s. Thus, it is presumed that smoking is unlikely to be a major contributor to the rising RCC incidence trends observed in this country.
1.2.2 Hypertension
The prevalence of hypertension in US adults has remained quite stable since the 1990s [13]. Over this period, there has been considerable improvement in awareness and control of the disease, and this may translate into benefits of reduced comorbidity for hypertensive patients in the future. Hypertension is a confirmed risk factor for development of RCC. Although hypertension is highly correlated with obesity, sufficient data supports its independent effect. A cohort study found that elevated systolic and diastolic blood pressure may predispose to development of RCC in a clear dose–response manner [14, 15]. A meta-analysis of 18 studies showed a significant 1.6-fold increased risk of RCC associated with hypertension [16]. It is difficult to isolate the contribution of antihypertensive medication use from history of hypertension because of the solid correlation between the two. Many observational studies have reported the hazardous impact on kidney cancer risk by taking antihypertensive medicine [16–18]. However, results from other studies indicated that instead of medication use, the excess risk is attributable to history of hypertension. For example, both systolic and diastolic blood pressure were associated with elevated risk of RCC when the analyses were restricted to non-antihypertensive medication users [19]. Clinical trials also showed no effect of taking antihypertensive medicine on cancer incidence [20]. The biological mechanisms that relate hypertension to RCC are still not fully understood. The renin–angiotensin system (RAS) plays an essential role in regulation of blood pressure and convincing evidence indicates that angiotensin II (Ang II) is involved in processes of cell proliferation, migration, angiogenesis, and inflammation [21]. Interestingly, slower development of tumor was observed in a xenograft mouse model of RCC when treated with captopril, an angiotensin-converting enzyme (ACE) inhibitor [22]. Furthermore, it is hypothesized that renal injury and metabolic or functional changes resulting from hypertension-induced chronic renal hypoxia and lipid peroxidation may subsequently raise renal susceptibility to carcinogens.
1.2.3 Energy Balance
1.2.3.1 Obesity
More than two thirds of US adults are overweight or obese [23]. Obesity has been linked to many chronic diseases including cancer. The association of obesity and breast cancer (postmenopausal), endometrial cancer, colon cancer, esophageal cancer, pancreatic cancer, and kidney cancer are well established [24, 25]. In the United States, about 40 % of RCCs are estimated to be attributable to being obese/overweight [5]. Interestingly, the effect of obesity has been reported to be specific for clear cell and chromophobe RCC histologies [26]. Although misclassification remains an inevitable limitation, body mass index (BMI) is widely accepted and used as a measure of obesity. Meta-analysis of prospective cohort studies has showed that each 5-unit increase in BMI contributes an approximate 24–34 % increase in risk of RCC for men and women [27]. From a statistical point of view, the effect size did not vary across ethnic groups, particularly in men. One interesting debate is at which period of life does being obese contribute most to the disease (i.e., childhood, adolescent, young adulthood, mid-age adulthood). One cohort study with more than 120,000 participants found association with BMI at baseline (aged 55–69 years) but not with BMI at age 20 years [28]. However, several studies have found that high BMI in adolescents may also confer excess risk [29–31]. These adolescent cohorts collected anthropometric measurements at baseline [29, 30]. BMI calculated from measured weight in the adolescent cohorts was more accurate than BMI calculated from recalled weight at their early ages by participants who are enrolled in most of the adult cohorts. It may provide more convincing evidence that obesity in early ages is associated with RCC risk.
Weight/BMI gain during adulthood is considered to be another risk factor for RCC that is independent of BMI per se, as reported in previous observational studies [1, 28, 31, 32]. The relationship is still inconclusive mainly due to confounding from obesity and inconsistent results. Similarly, weight/BMI gain relied on recalls from participants at enrollment for most of the studies that the misclassification remains an issue. A longitudinal study with multiple assessments of weight and adequate adjustment of BMI would be valuable to disentangle the complicated interplay. However, such a study is difficult to conduct in practice. Likewise, the effects of other related factors, such as weight cycling and waist–hip ratio, are also tightly correlated with BMI so their independent influences are difficult to assess.
Various mechanisms have been proposed for the association between obesity and cancer. Increased mass of adipose tissue is not the only change that occurs in obese subjects. Levels of circulating adipokines and sexual hormones are altered as well. Additionally, it appears that higher concentrations of insulin and increased levels of insulin resistance were observed in subjects with abnormal BMI. Extensive data has linked leptin, adiponectin (inversely), hyperinsulinemia, as well as insulin-like growth factor (IGF) and its binding proteins to tumorigenesis through stimulation of cell proliferation, angiogenesis, and inhibition of apoptosis [24, 33]. Elevated circulating levels of pro-inflammatory cytokines, such as interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α), chronic tissue hypoxia, lipid peroxidation, and increased oxidative stress have also been hypothesized to connect obesity to cancer. A limited number of studies have investigated the underlying mechanisms relating obesity to RCC. Two case-control studies showed contrasting results of association between serum leptin and RCC risk [34, 35]. Similarly, the association of adiponectin with RCC remains inconclusive [34, 36, 37]. Reverse causation may affect the validity of retrospectively designed studies. Prospective cohort studies with large sample size and nested case-control studies are warranted for the confirmation of previous findings.
1.2.3.2 Diet and Alcohol Consumption
On average, one third of cancers are estimated to be attributable to diet and nutrition [38]. Western diets are criticized for high intake of calories, meat and saturated fats, and their link to obesity and other chronic diseases. Incidence rates of RCC in the American Asian population are more than twofold that observed in Asian countries [2, 4]. Apparently, acculturation, such as changes in diet, is more likely to lead to the discrepancy rather than alterations in genetics.
High energy intake is a potential risk factor for RCC. However, due to its tight correlation with BMI, the association is still controversial. Several studies that evaluated energy intake and its association with RCC risk reported inconsistent results, even with adequate adjustment of BMI and other confounders [28, 39]. Various components of diet and nutrients in foods have been assessed for RCC risk. Although one recent study restricted to Caucasian male smokers showed no evidence of association [40], high fruit and vegetable consumption was associated with a >30 % reduction of RCC risk in a pooled analysis of 13 cohort studies [41]. It is interesting to further isolate the specific vegetable or fruit that drives the association. Suggestive inverse associations were found for highest quartile of cruciferous vegetables and whole citrus fruits consumption when compared to the lowest category [42, 43]. Furthermore, high intake of dietary fiber was also inversely associated with RCC risk in a previous study [43]. Antioxidants may play a protective role in cancer development, although the reported associations of antioxidant nutrients including carotenoids and vitamins A, C, and E are inconsistent [41, 42, 44, 45]. Another speculation is that B vitamins and other components of the one-carbon metabolisms pathway which are important for DNA repair mechanisms in the body could partially explain the inverse association for fruits and vegetables. One cohort study reported that higher vitamin B6 level in plasma is in relation to both RCC incidence and survival [46].
Meat consumption may confer increased risk. A meta-analysis of cohort studies found that intake of fat and protein or their subtypes were not associated with RCC risk after adjusting for BMI, fruit and vegetable intake, and alcohol consumption [47]. In contrast, more recent studies have suggested a link and underscored the role of heterocyclic amines (HCAs), and polycyclic aromatic hydrocarbons (PAHs) found in meat cooked at high temperature [48, 49]. The intake of dietary acrylamide which is concentrated in baked and fried carbohydrate-rich foods was reported to confer a 60 % increased risk of RCC in a case cohort study in the Netherlands [50].
Regarding alcohol, moderate consumption was reported to be inversely associated with the risk of RCC [51]. A 28 % reduction in risk was observed in people who consumed >15 g per day of alcohol (equivalent to 1–2 drinks/day). No further reduction was observed for additional increases in consumption [52]. A recent large prospective cohort study has found a dose–response effect for alcohol consumption [53]. The protective effect is likely to be attributable to improved insulin sensitivity and antioxidant compounds contained in alcoholic beverages. The associations with intake of other beverages, such as coffee, milk, and tea, are inconsistent [1, 54].
Due to the high dimensions of nutrition data and collinearity among food items/nutrients, PCA (principal component analysis) and factor analysis are adopted for the purpose of data reduction. Interactions between different food items/nutrients are taken into account in this type of analysis. Suggestive evidence showed that patterns of “alcohol drinking” were inversely associated with RCC risk [55], and high-calorie, high-protein, and high-fat food patterns conferred an increased risk [56].
1.2.3.3 Physical Activity
Physical activity accounts for a large proportion of daily energy expenditure, which affects the status of energy balance of individuals. Most studies have focused on non-occupational or leisure-time physical activity and its association with cancer risk. Reduced RCC risk was reported by a number of observational studies for people with high level of physical activity [1]. Meta-analysis using the random effect model found an inverse association when comparing high levels to low levels of physical activity [57]. The heterogeneity seen across studies in the meta-analysis may be partially attributable to the differences in measures of physical activity between studies that included leisure-time physical activity during the past year, frequency of physical activity during a certain period, hours spent on physical activity per day or per week, and metabolic equivalent task (MET) values. Although such differences were not statistically significant, the magnitude of estimations based on frequency or duration of physical activity appeared to be stronger than that estimated by METs or qualitative physical activity. Occupational physical activity studies are scarce and have mixed results [58, 59]. The beneficial effect of physical activity on RCC is likely to be mediated by reduction in body weight/ BMI, lowered blood pressure, changes to adipokines, alleviation of insulin resistance, and an improved profile of inflammation and oxidative stress.
1.2.4 Other Medical, Occupational, and Environmental Factors
Type II diabetes mellitus (DM) could be a risk factor for RCC independent of obesity/BMI. A meta-analysis that adjusted for obesity/BMI, alcohol consumption, and smoking has shown that a history of type II DM significantly confers higher predisposition of RCC [60]. However, only a few of the individual studies were further adjusted for history of hypertension that residual confounding may distort the association. In women, parity increases disease risk when compared to nulliparous, and a dose–response effect for parity number was significant in a meta-analysis [61]. Inflammation and elevated oxidative stress through pregnancy-induced physiologic changes, pregnancy-associated weight gain, and high levels of circulating estrogens have all been hypothesized as underlying mechanisms. There are other reported risk factors with inconclusive associations, such as end-stage renal disease, long-term hemodialysis, acquired renal cystic disease, use of statin and aspirin, and occupational and other environmental exposures [1, 62].
1.3 Genetic Susceptibility
1.3.1 Hereditary Kidney Cancer Syndrome
Compelling evidence supports genetic susceptibility to RCC. The risk of RCC is two to three times higher in individuals who have first-degree relatives with kidney cancer [63]. In addition, familial aggregation is seen in approximately 3 % of kidney cancer patients with inherited kidney cancer syndromes. Of these, the von Hippel–Lindau syndrome (VHL) is by far the most commonly recognized familial cancer syndrome associated with kidney cancer [64]. Other major familial kidney cancer syndromes include hereditary papillary renal cell carcinoma (HPRC), hereditary leiomyomatosis renal cell carcinoma (HLRCC), and Birt–Hogg–Dubé syndrome (BHD) [65], which are caused by mutations in the c-Met proto-oncogene, FH (fumarate hydratase), and FLCN (folliculin), respectively. Interestingly, VHL, FH, c-Met, and FLCN are all involved in the pathways that respond to nutrient stimulation and cell metabolism, indicating kidney cancer is a metabolic disease [66].
1.3.2 Candidate Gene Approach
Most of the early candidate gene studies involved small numbers of cases and controls, and very few of the initially reported positive susceptibility alleles have been replicated in subsequent validation studies [67]. With regard to RCC, candidate gene studies reported many positive associations with single nucleotide polymorphisms (SNPs) in genes involved in xenobiotic metabolism, DNA repair, cell growth/apoptosis, inflammation, and other pathways [1]. None of the previously reported candidate SNPs has been replicated in large independent studies.
1.3.3 Genome-Wide Association Study (GWAS)
The advent of GWAS in recent years has revolutionized the study of cancer association. Unlike the hypothesis-driven candidate gene approach, GWAS is a discovery-driven, agnostic approach that does not depend on prior knowledge of SNPs and genes. It thoroughly screens up to millions of common SNPs across the entire genome. Due to the multiple testing of SNPs in the screening phase, to increase validity of the results, stringent Bonferroni correction (P-value < 5×10−8) and multistage follow-up validations with large sample sizes are required.
Three recent GWASs identified four novel genetic susceptibility loci that mapped to 2p21 (EPAS1), 11q13.3 (a CCND1 transcriptional-enhancer site), 12p11.23 (ITPR2), and 2q22.3 (ZEB2) [68–70]. The observed effect size of each genetic locus is relatively small, which is expected for common variants. Two validated SNPs are located in intron 1 of EPAS1 (endothelial PAS domain-containing protein 1) on chromosome 2p21. However, the putative function of these SNPs is still unclear. EPAS1 encodes HIF-2α, which is a biologically plausible causal gene in the VHL/HIF pathway. Thus, GWAS results provide further support for the involvement of EPAS1 in RCC etiology. Interestingly, although the SNP found in 11q13.3 is not close to any gene with known function (>50 kb), the locus is hypothesized to be a transcriptional-enhancer site of CCND1 (encoding cyclin D1) [71]. Another locus maps to ITPR2 (inositol 1,4,5-trisphosphate receptor, type 2) on 12p11.23. Interestingly, the same SNP has also been identified as being associated with waist–hip ratio in another GWAS [72]. Finally, the locus on 2q22.3 is mapped to ZEB2 (zinc finger E-box-binding homeobox 2) that functions as a DNA-binding transcriptional repressor. Future pooled analysis of GWAS data with larger sample size will undoubtedly identify additional common RCC susceptibility SNPs.
1.3.4 Next-Generation Sequencing
The emergence of next-generation sequencing (NGS) provides a unique opportunity to discover rare variants that may explain some extent of the missing heritability in complex diseases. However, current NGS studies of kidney cancer have been mostly focused on profiling somatic mutations, while large-scale NGS studies using germline DNA have not been conducted. For example, a whole-exome sequencing (WES) study of tumor tissues has identified the SWI/SNF chromatin remodeling complex gene PBRM1 (polybromo 1) as the second most frequently mutated gene in ccRCC [73]. Another study reported that the tumor suppressor gene BAP1 (BRCA1 associated protein-1) could be used to define a new class of RCC [74]. With more than 400 tumor samples, The Cancer Genome Atlas (TCGA) identified 19 significantly mutated genes. Integrative analyses highlighted the importance of previously well-known pathways such as the VHL/HIF pathway, chromatin remodeling pathway, and PI3K/AKT/mTOR pathway [75]. Future NGS study using germline DNA may focus on identifying rare mutations, which can play critical role(s) in kidney cancer development.
1.3.5 Intermediate Phenotypic Assays for RCC Susceptibility
Intermediate phenotypic biomarkers have the advantage of measuring aggregated effects of genetic variations and have larger effect size than individual SNPs. The suboptimal DNA damage/repair capacity in PBLs that were challenged with different mutagens was shown to be associated with increased risk of RCC [11, 12, 76]. One study found BPDE (benzo[α]pyrene diol epoxide) induced lymphocytic chromosome 3p deletion was associated with RCC risk [11]. Two other studies used comet assays to show that the high sensitivity of PBLs to NNK (nicotine-derived nitrosamine ketone)- and BPDE-induced DNA damage was associated with increased risk of RCC [12, 76]. Another interesting phenotypic biomarker is telomere length. Telomeres protect chromosomes from degradation and end-to-end fusion. Short telomere length in PBLs was associated with increased risk of a variety of cancers [77]. However, the association in RCC is inconsistent. Two case-control studies found that shorter telomeres in PBLs to be associated with an increased risk of RCC [78, 79] and one prospective nested case-control study failed to confirm the association [80]. Mitochondrial DNA (mtDNA) copy number is recently attracting great interest as a potential cancer susceptibility marker. Decreased mtDNA copy number was shown to be associated with multiple types of cancer. In two studies, lower mtDNA copy number in PBLs conferred an increased risk of RCC [81, 82]. By contrast, a recently published prospective study showed that a high copy number of mtDNA increased risk [83].
Related posts:
 Predictive and Prognostic Markers in Metastatic Renal Cell Carcinoma
Mammalian Targets of Rapamycin Inhibitors: Temsirolimus and Everolimus
Predictive and Prognostic Markers in Metastatic Renal Cell Carcinoma
Mammalian Targets of Rapamycin Inhibitors: Temsirolimus and Everolimus
 Forthcoming Drugs for Metastatic Renal Cell Carcinoma Therapy
Forthcoming Drugs for Metastatic Renal Cell Carcinoma Therapy
 Optimization of Therapy by Pharmacokinetic–Pharmacodynamic Analyses
Optimization of Therapy by Pharmacokinetic–Pharmacodynamic Analyses
 Treatment Overview
Treatment Overview
 Hereditary Renal Cell Carcinoma
Hereditary Renal Cell Carcinoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


