Diffuse astrocytic and oligodendroglial tumors
ICD-O codes
Diffuse astrocytoma, IDH-mutant
9400/3
Gemistocytic astrocytoma, IDH-mutant
9411/3
Diffuse astrocytoma, IDH-wildtype
9400/3
Diffuse astrocytoma, NOS
9400/3
Anaplastic astrocytoma, IDH-mutant
9401/3
Anaplastic astrocytoma, IDH-wildtype
9401/3
Anaplastic astrocytoma, NOS
9401/3
Glioblastoma, IDH-wildtype
9440/3
Giant cell glioblastoma
9441/3
Gliosarcoma
9442/3
Epithelioid glioblastoma
9440/3
Glioblastoma, IDH-mutant
9445/3a
Glioblastoma, NOS
9440/3
Diffuse midline glioma, H3 K27M-mutant
9385/3a
Oligodendroglioma, IDH-mutant and 1p/19q-codeleted
9450/3
Oligodendroglioma, NOS
9450/3
Anaplastic oligodendroglioma, IDH-mutant and 1p/19q-codeleted
9451/3
Anaplastic oligodendroglioma, NOS
9451/3
Oligoastrocytoma, NOS
9382/3
Anaplastic oligoastrocytoma, NOS
9382/3
Table 2.2
Grading of selected diffuse astrocytic and oligodendroglial tumors according to the 2016 WHO Classification, adapted from Louis et al. 2016 [2]
Selected diffuse astrocytic and oligodendroglial tumors | Grade |
|---|---|
Diffuse astrocytoma, IDH-mutant | II |
Anaplastic astrocytoma, IDH-mutant | III |
Glioblastoma, IDH-wildtype | IV |
Glioblastoma, IDH-mutant | IV |
Diffuse midline glioma, H3 K27M-mutant | IV |
Oligodendroglioma, IDH-mutant and 1p/19q-codeleted | II |
Anaplastic oligodendroglioma, IDH-mutant and 1p/19q-codeleted | III |
In this new classification, the diffuse gliomas include the WHO grade II and grade III astrocytic tumors, the grade II and III oligodendrogliomas, the grade IV glioblastomas, as well as the related diffuse gliomas of childhood. This approach leaves those astrocytomas that have a more circumscribed growth pattern, lack IDH gene family alterations and frequently have BRAF alterations (pilocytic astrocytoma, pleomorphic xanthastrocytoma) or TSC1/TSC2 mutations (subependymal giant cell astrocytoma), distinct from the diffuse gliomas. In other words, diffuse astrocytoma and oligodendrogliomas are now nosologically more similar than diffuse astrocytoma and pilocytic astrocytoma are [2].
Furthermore, two diffuse astrocytoma variants have been deleted from the 2007 WHO Classification: protoplasmic astrocytoma, and fibrillary astrocytoma, since this diagnosis overlaps nearly entirely with the standard diffuse astrocytoma. As a result, only gemistocytic astrocytoma remains as a distinct variant of diffuse astrocytoma, IDH-mutant.
The diagnosis of oligodendroglioma and anaplastic oligodendroglioma requires the demonstration of both an IDH gene family mutation and combined whole-arm losses of 1p and 19q (1p/19q codeletion). In the absence of positive mutant R132H IDH1 immunohistochemistry, sequencing of IDH1 codon 132 and IDH2 codon 172 is recommended. In the absence of testing capabilities or in the setting of inconclusive genetic results, a histologically typical oligodendroglioma should be diagnosed as NOS.
In the 2016 CNS WHO Classification, the diagnosis of oligoastrocytoma is strongly discouraged. Nearly all tumors with histological features suggesting both an astrocytic and an oligodendroglial component can be classified as either astrocytoma or oligodendroglioma using genetic testing. The diagnoses of WHO grade II oligoastrocytoma and WHO grade III anaplastic oligoastrocytoma are, therefore, assigned NOS designations, indicating that they can only be made in the absence of appropriate diagnostic molecular testing [2].
Thus in 2016, strictly speaking, DLGGs include (with the corresponding ICD-O codes):
- 1)
Diffuse astrocytoma, IDH-mutant (9400/3),
- 2)
Gemistocytic astrocytoma, IDH-mutant—which is a variant of the first one (9411/3)
- 3)
Diffuse astrocytoma, IDH-wildtype (9400/3),
- 4)
Diffuse astrocytoma, NOS (9400/3),
- 5)
Oligodendroglioma, IDH-mutant and 1p/19q-codeleted (9450/3),
- 6)
Oligodendroglioma, NOS (9450/3),
- 7)
Oligoastrocytoma, NOS (9382/3).
From an epidemiological point of view, three important issues should be noted regarding this new classification:
First, this new classification underscores the link between DLGG, diffuse anaplastic glioma (DAG), and even glioblastoma IDH mutant. The question of the spontaneous occurrence of DAG or the constant occurrence of DAG from initially DLGG is currently not resolved, but many factors are in favor of the transformation of DLGG. This point is important in epidemiology, because it may change the values of incidence and prevalence of these tumors.
Secondly, as it was in the previous classification, ICD-O codes are not enough specific to describe all DLGG subtypes (because some DLGG subtypes have the same code). That is why some brain tumor databases (as the French Brain Tumor DataBase) suggest recording both the ICD-O code and the histological type and subtyping for each entity.
Thirdly, because the 2016 WHO Classification is new, no data are yet available in brain tumor registries or in population studies. Only specific studies that include molecular parameters are available (see the chapter by Jacobs et al. on “Molecular Epidemiology of DLGG”).
So, in the present chapter, we will only describe epidemiology for DLGG defined by the previous WHO classification.
Moreover, specific epidemiological publications for DLGG are rare. However, it is possible to obtain epidemiological data concerning DLGG by selecting some articles referring to all primary central nervous system tumors (PCNSTs), neuroepithelial tumors, gliomas, or even to low grade gliomas (LGGs). These publications can come from registry works, or from consortium studies. The main difficulties are that definitions, referring histology or topography coding could vary with time, countries, and people involved in.
In the first part of this chapter, we will focus on PCNST generalities and definitions, then we will discuss the proportion of DLGG among PCNST, sex ratio and median ages at diagnosis for DLGG, and we will show few surgical data for gliomas in France. We will finish the first part by explaining why the term of DLGG is more appropriate than the term of LGG.
The second part will present data concerning incidence, survival and prevalence for DLGG.
The third part will summarize prognostic factors for DLGG, knowing that they will be detailed in other chapters of this book.
The last part will introduce new methodologies: (1) for improving clinical epidemiology (to benefit from a best evaluation of oncological care on survival and on quality of life for DLGG patients), (2) for looking for etiologies for these tumors, because until now, except in very few cases, the causes of DLGG are not known, and (3) for early detection of DLGGs.
2.2 PCNST and DLGG: Generalities
2.2.1 PCNST Generalities and Definitions
PCNSTs represent a complex heterogeneous group of pathological entities that may be benign, malignant or of unpredictable evolution [1–10]. These tumors represent a major public health problem [11]. The term “primary brain tumor” has been defined in numerous ways in the literature. The primary difficulty in building a tumor registry and/or a data base is to define the type of tumor to be recorded.
Recent publications [12–20], the classification system of the World Health Organization [1, 2, 6], and the European recommendations for coding tumors of the brain and CNS [21] include all primary benign and malignant tumors located in the CNS, including its envelopes and the beginning of the nerves localized in the skull and spine. PCNSTs include tumors of neuroepithelial tissue (gliomas and all other neuroepithelial tumors), tumors of the meninges (meningiomas, mesenchymal tumors and other tumors of the meninges), tumors of the cranial and paraspinal nerves, lymphomas and hematopoietic neoplasms, and others.
By definition, this excludes metastatic tumors. But even now, there are still discrepancies concerning the inclusion of lymphoma, pituitary and pineal glands, and olfactory tumors of the nasal cavity, in the different databases and registries. It is important to note that some institutions account for primary malignant tumors, only.
Coding systems have been introduced as an indispensable interface between pathologists and cancer registries. The international classification of diseases for oncology (ICD-O) was established more than 30 years ago. It assures that histopathologically stratified population-based incidence and mortality data become available for epidemiological and oncological studies. The histology (morphology) code is increasingly complemented by genetic characterization of human neoplasms. The ICD-O histology codes have been adopted by the systematized nomenclature of medicine (SNOMED), issued by the College of American Pathologists. ICD-0-3 and SNOMED codes are available in Louis et al. [1, 2] and in Rigau et al. [19]. All glioma morphology codes are shown here (see Table 2.3) [23].
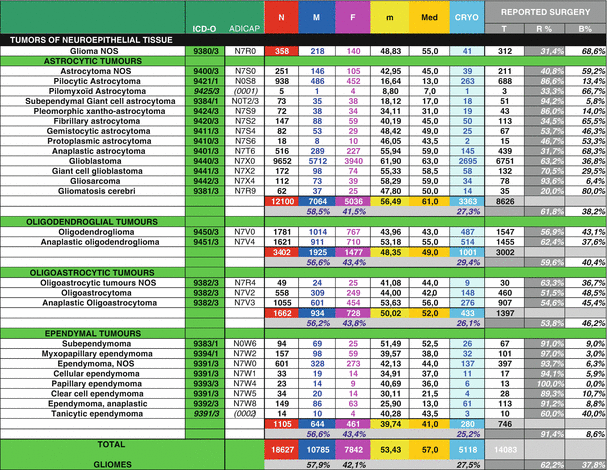
Table 2.3
Histological repartition of the 18,627 glioma cases with clinical and surgical data from French Brain Tumor Database, period 2004–2009, adapted from Zouaoui et al. [22]
The ICD-O topography codes largely correspond to those of the tenth edition of the International statistical classification of diseases, injuries and causes of death (ICD-10) of the WHO. ICD-O-3 topography codes include: brain (C71.0–C71.9), meninges (C70.0–C70.9), spinal cord, cauda equina, cranial nerves, and other parts of the central nervous system (C72.0–C72.9) [24]. Now, many registries use these codes, but some registries still record malignant tumors only [25]. Some registries (e.g., Central Brain Tumor Registry of the United States –CBTRUS-) include more topography codes as pituitary and pineal glands (C75.1–C75.3), and olfactory tumors of the nasal cavity [C30.0 (9522–9523)].
Another challenge for registries and databases is to record and to detail all cases of defined tumors (i.e., all histological types and all histological subtypes, when it is possible).
2.2.2 Proportion of DLGGs Among PCNSTs and Gliomas
We present the proportion of each major type of PCNST and the distribution of DLGG in the French Brain Tumor DataBase (FBTDB) for the 2006–2011 period (Fig. 2.1a–c). We point out (1) FBTDB records cases with histological confirmation only, knowing that usually, registries record the cases with and without histological confirmation, and (2) the distribution of the different subtypes of PCNST is similar in France and in the USA, except for oligodendroglial tumors (see [19]). Recently, most studies (e.g., [26, 27]) have reported a recent increase of oligodendroglial tumors in comparison to astrocytic tumors; however, French neuropathologists are more influenced by the classification proposed by Daumas-Duport et al. [28] than American neuropathologists.
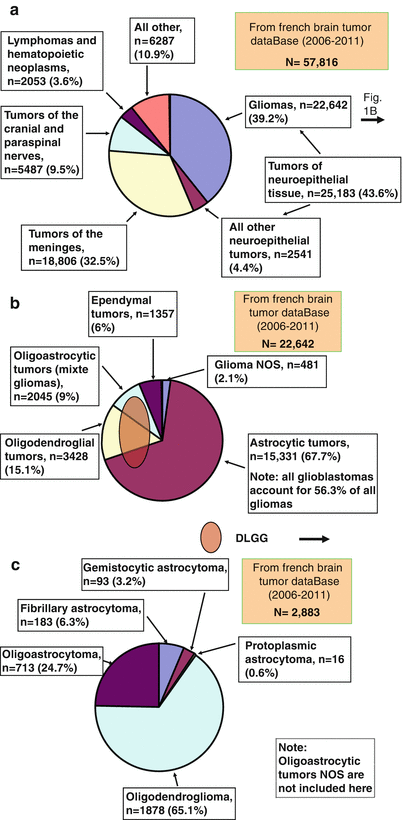
Fig. 2.1
(a–c) Data from French Brain Tumor DataBase (2006–2011). (a) Distribution of all primary central nervous system tumors by main histological types. (b) Distribution of all gliomas (all sites: included supratentorial, infratentorial and spinal cord) by main histological subtypes. (c) Distribution of all diffuse low grade gliomas (all sites: included supratentorial, infratentorial and spinal cord) by all histological subtypes, excepted oligoastrocytic tumors NOS. Abbreviations: DLGG diffuse low grade glioma, NOS not otherwise specified
We could see here the difficulties to compare data from different institutions when histological diagnosis is not completely reproducible [29].
An additional difficulty to know the overall proportion of DLGG is that (1) some institutions use terminology such as “low grade gliomas” (LGG, see below) and (2) when some information are not detailed enough, registries include some cases in astrocytoma not otherwise specified (NOS) or in glioma NOS, even in unspecified neoplasm.
However, we can consider that the overall DLGG represents about 13–16% of all gliomas.
2.2.3 Sex Ratio, Median Age at Diagnosis and Few Surgical Data for DLGG
The number of males and females and the median age at diagnosis (MAD) for DLGG in FBTDB (period: 2004–2009) [23] are shown in Table 2.3. Sex ratios, MAD in CBTRUS [15, 17] and in FBTDB are shown in Table 2.4. Sex ratio (male/female) for all DLGG is very similar in CBTRUS (1.33) and in FBTDB (1.32). It is the same for MAD for each sub type of DLGG. One can hypothesis that these similarities across the Atlantic could argue for genetic components of these tumors. To be rigorous about classification system, we can notice (1) gemistocytic astrocytoma were not included in DLGG in 2009 and 2012 CBTRUS reports (probably, because gemistocytic astrocytomas are more aggressive tumors, CBTRUS counted gemistocytic astrocytomas with anaplastic astrocytomas [note: 2015 CBTRUS report includes gemistocytic astrocytomas in diffuse astrocytomas, as in the 2007 WHO Classification] (2) CBTRUS does not separate oligoastrocytomas from anaplastic oligoastrocytomas, because these tumors have the same SNOMED code (9382/3) (see Tables 2.1 and 2.3).
Another important thing to notice when one speaks of MAD is how diagnosis is defined. When surgery is performed, registries and the large majority of databases use usually the date of the histological diagnosis. If we refer to clinical studies, some of them specify the age of radiological diagnosis, or sometimes, the age at first symptom. For example, the “Reseau d’étude des gliomes” (REG, a French consortium that studies DLGG) considered several starting points for their analyses: the date of available radiological diagnosis (which did not differ significantly from the date of clinical onset) and the date of first treatment. Among 1091 patients (collected at adult age), it was noted that median age (MA) at discovery was 37 years old (rage: 4–75) and the MA at first treatment was 44 years old (range: 18–76) [30]. Probably, this discrepancy would be decreased in the future, because clinicians seem to treat DLGG earlier than in the past.
Surgical data concerning DLGG are rare in population studies. Table 2.3 shows the percentages of resection versus biopsy for 14,083 gliomas and all subtypes (included DLGG) (FBTDB, collected data from 2004 to 2009). Of the 54 institutions that participated, the proportion of resection versus biopsy varied considerably from one institution to another (data not shown). We can also note that at least 27% of all glial tumors were cryopreserved (see [19, 23]). This is very important for future biological studies.
2.2.4 The Term of DLGG Is More Appropriate Than the Term of LGG
For many years, epidemiological studies have investigated all gliomas or even all PCNST. More recently, the term “low grade glioma (LGG)” has been introduced. LGG is probably used as a practical definition being quite simply in opposition to the term “high grade glioma (HGG)”. LGGs are slow growing, intrinsic lesions that contain glioma cells. Referring to the WHO classification of tumors of the CNS by Louis et al. [1], LGG may be defined by grade I (GI) and grade II (GII) gliomas and includes subependymal giant cell astrocytoma (GI), pilocytic astrocytoma (GI), pilomyxoid astrocytoma (GII), diffuse astrocytoma (GII), pleomorphic xanthoastrocytoma (GII), oligodendroglioma (GII), oligoastrocytoma (GII), subependymoma (GI), mixopapillary ependymoma (GI) and ependymoma (GII). Some authors include gangliomas, desmoplastic gangliomas [31], and even dysembryoplastic neuroepithelial tumors (DNETs) [32]. However, many studies on LGG exclude ependymomas and refer to astrocytic and/or oligodendrocytic tumors only. Furthermore, some pediatric studies mainly focus on pilocytic astrocytic tumors, and some adult studies often focus on GII astrocytic and/or oligodendrocytic tumors only.
Here, the term “diffuse low grade gliomas (DLGG)” includes grade II gliomas for diffuse astrocytomas, oligodendrogliomas and oligoastocytomas (mixed gliomas). This term is much more precise—because in essence it excludes well delineated grade I and II gliomas, and ependymomas with a different natural history. One benefit of this term is also to ignore the difficulties to differentiate an oligoastrocytoma from an astrocytoma or an oligodendroglioma. Moreover, even if heterogeneities are present in the GII glioma group of diffuse astrocytomas, oligodendrogliomas and oligoastocytomas, these tumors are often studied together. Usually they concern middle-aged adults with professional activities. The main clinical presentation is epilepsy with or without mild cognitive disorders. Focal deficit and/or raised intracranial pressure are possible but very infrequent. And the majority of these tumors have typical imaging characteristics on MRI as non-enhancing infiltrative lesions involving the white matter and frequently extending to the cortical surface [33]. Moreover, now the new WHO CNS Classification specifies the group of diffuse astrocytic and oligodendroglial tumors [2], Tables 2.1 and 2.2.
2.3 Incidence, Survival and Prevalence for DLGG
Epidemiological data for DLGG are patchy. In 2016, we can consider that we have (1) good incidence data in some countries, (2) some interesting elements regarding survival, (3) little items on the prevalence.
2.3.1 Incidence
As mentioned previously, incidence data could vary from countries because the coding rules are not exactly the same all over the world. For example, referring to the 2007 WHO Classification, diffuse astrocytoma include fibrillary astrocytoma, gemistocytic astrocytoma, and protoplasmic astrocytoma, but some registries include astrocytoma NOS also. Classically, registries record cases with and without histological confirmation. It means that a number of cases of tumor are diagnosed by radiology, or even by clinical data only, or even by death certificate only. This is why the author decides to show data from (1) registries (e.g., Central Brain Tumor Registry of the United States—CBTRUS, and Austrian Brain Tumor Registry—ABTR) and (2) from histological population-based study (e.g., French Brain Tumor DataBase—FBTDB) that include all cases with histological validation only (Table 2.5). Histological population-based studies are more accurate but underestimate the true incidence [34–36]. Some other publications (e, g., [20, 37, 38]) present incidence data on DLGG, and one recent publication summarizes incidence data of glioma and some sub-types in different countries [39].
Table 2.5
Comparison of the incidence rates (IR) of DLGGs and some PCNSTs (per 100,000) from USA registry (CBTRUS), Austrian registry (ABTR), and FBTDB
Registry or population-based study | IR adjusted on the USA population | IR adjusted on the worldwide population | IR adjusted on the Europe population | IR adjusted on the French population |
|---|---|---|---|---|
CBTRUSa 2008–2012 [18] | ||||
Tumors of neuroepithelial tissue | 6.62 | |||
Diffuse astrocytomab | 0.53 | |||
Oligodendroglioma | 0.25 | |||
Oligoastrocytic tumorsc | 0.20 | |||
Totalb,c | 0.98 | |||
FBTDBd 2006–2011 | ||||
Tumors of neuroepithelial tissue | 6.160 | 5.507 | 6.343 | 6.771 |
Gliomas | 5.418 | 4.623 | 5.587 | 6.088 |
Fibrillary astrocytoma | 0.048 | 0.050 | 0.051 | 0.049 |
Gemistocytic astrocytoma | 0.024 | 0.021 | 0.025 | 0.025 |
Protoplasmic astrocytoma | 0.004 | 0.004 | 0.004 | 0.004 |
Oligodendroglioma | 0.501 | 0.437 | 0.502 | 0.505 |
Oligoastrocytoma | 0.191 | 0.171 | 0.192 | 0.192 |
Totale | 0.768 | 0.683 | 0.774 | 0.775 |
FBTDBd 2006–2009 [33] | ||||
All diffuse grade II gliomasf | 0.83 | 0.76 | 0.86 | 0.85 |
ABTRa 2005 [14] | ||||
Tumors of neuroepithelial tissue | 7.26 | |||
Gliomas | 6.6 | |||
Diffuse astrocytomab | 0.75 | |||
Oligodendroglioma | 0.20 | |||
Oligoastrocytoma | 0.27 | |||
Totalb | 1.22 | |||
When we want to compare the incidence of one specific tumor from one country to another, another issue to consider is the differences in the two populations. Crude rates, whether they represent incidence, mortality, morbidity or other health events, are summary measures of the experience of populations that facilitate this comparative analysis. However, the comparison of crude rates can sometimes be inadequate, particularly when the population structures are not comparable for factors such as age, sex or socioeconomic level. For example, median age at diagnosis of glioblastoma is 64 years; because the age distribution is different in occidental word than in developing countries, we can not compare the crude rate of glioblastoma in US to the crude rate of glioblastoma in India. To enable international comparison, age-standardized rates from the FBTDB data were calculated with the USA, Worldwide, and Europe populations as references (Table 2.5).
Moreover, from epidemiological point of view, the difficulty in grading the diffuse gliomas in grade II vs. III by pathologists in some cases could modify the incidence of DLGG vs. DAG (e.g., microfoci in DLGG [40]). In the past, neurosurgeons gave only small samples to pathologists; now bigger samples of the tumor are analyzed by the pathologist, and pathologists have more sophisticated techniques. So we could imagine that incidence of DLGG would decrease slightly while incidence of DAG would increase slightly.
The 2016 WHO Classification integrates biology but the difficulties still persist in differentiating GII and GIII in some cases.
In occidental world, the incidence rate for all DLGG is between 0.6 and 1.3/100,000 person-years. So, we can hypothesize that the value of 1/100,000 person-years, or a little bit less, could be considered as a good approximation.
So, this first conclusion about incidence of about 1/100,000 person-years for DLGG in occidental world implies few comments and questions:
The variation of incidence between each subtype of DLGG seems more important than the variation of the overall incidence. It means that histological criteria between astrocytoma, oligodendroglioma and oligoastrocytoma are probably variable from one country (or one center) to another. Does the new WHO Classification will do better? We cannot answer yet.
What can explain the (relatively small) discrepancies between the overall incidence of DLGG from one country (or one area) to another, in the occidental world? Only methodology? Or real difference? We cannot answer yet. But we can notice that incidence of overall DLGG is higher (1) in men than in women (see sex ratio of M/F of about 1.3), and (2) in White than in Black in USA. CBTRUS data [15] give incidence rates (per 100,000 person-years, age-adjusted to the 2000 U.S. standard population): protoplasmic and fibrillary astrocytoma: 0.10, oligodendroglioma: 0.31, mixed glioma: 0.19; Male/Female: 0.13/0.08, 0.34/0.27, 0.24/0.16; White/Black: 0.12/0.04, 0.34/0.13, 0.21/0.08 for the same histology respectively. Furthermore, the FBTDB showed that the geographical distribution by region of the DLGG had significant differences, with higher incidence rates in Northeast and central parts of France [34].
From epidemiological point of view, two additional points need to be mentioned. First, the incidental findings on MRI are increasing with the use of MRI for many other clinical situations (brain traumas, headaches, neurologic disorders, etc.) and even for research purposes [41–43]. Secondly, DLGG patients have a long silent stage before to be symptomatic, and some teams discuss screening and preventive therapeutic management [44–46]. So, we can imagine that the incidence of DLGG could rise in the future.
2.3.2 Survival
In 2016, many interesting publications are available in the literature about gliomas survival. But unfortunately, most of them include a lot of different subtypes. Many publications analyze only clinical, or therapeutic, or biologic prognostic factors. So, it is very difficult to conclude precisely. Of course survival of DLGG is better than diffuse high grade gliomas, but DLGG is still an ultimately fatal disease.
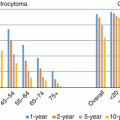
US survival rates for DLGG [diffuse astrocytoma (including fibrillary, gemistocytic, protoplasmic, and NOS astrocytomas), oligodendroglioma, and mixed glioma (including oligoastrocytoma grade II and III because they have the same SNOMED code) and selected glioma subtypes [i.e., anaplastic oligodendroglioma, anaplastic astrocytoma, glioblastoma, and glioma malignant NOS] for comparison, are presented in Table 2.6 (adapted from 2015 CBTRUS report [18], period 1995–2012). The estimated 5-/10-year relative survival rates for diffuse astrocytomas and oligodendrogliomas are 47.9/37.6 and 79.8/64.0%, respectively. Three points could be noted: (1) the estimated 5- and 10-year relative survival rates for mixed gliomas (included oligostrocytomas and anaplastic oligoastrocytomas) are 62.0/47.8%, respectively; (2) the number of glioma NOS is particularly high and (3) survival of glioma NOS is not so poor as glioblastoma survival.
Table 2.6
One-, Two-, Three-, Four-, Five-, and Ten-year relative survival ratesa,b for diffuse low grade gliomas, anaplastic astrocytoma, anaplastic oligodendroglioma, glioblastoma, and glioma malignant NOS, SEER 18 registries, 1995–2012c, from 2015 CBTRUS report [18]
Histology | Nd | 1-Yr | 2-Yr | 3-Yr | 4-Yr | 5-Yr | 10-Yr |
|---|---|---|---|---|---|---|---|
Diffuse astrocytomae | 6635 | 72.2% | 61.5% | 55.4% | 51.2% | 47.9% | 37.6% |
Oligodendroglioma | 3602 | 93.9% | 89.5% | 86.2% | 82.9% | 79.8% | 64.0% |
Mixed gliomaf | 2130 | 87.6% | 77.9% | 71.4% | 66.1% | 62.0% | 47.8% |
Anaplastic astrocytoma | 4101 | 62.1% | 44.0% | 35.70% | 31.2% | 27.9% | 19.8% |
Anaplastic oligodendroglioma | 1441 | 81.5% | 68.9% | 62.4% | 57.0% | 52.5% | 38.9% |
Glioblastoma | 33,204 | 37.2% | 15.2% | 8.8% | 6.3% | 5.1% | 2.6% |
Glioma malignant, NOS | 4717 | 63.2% | 52.7% | 49.3% | 47.6% | 46.1% | 41.3% |
In the population-based study of the Canton of Zurich, Switzerland (N = 122 DLGG, period 1980–1994), the survival rate (mean follow-up 7.5 ± 4.8 years) was highest for patients with oligodendroglioma (78% at 5 years, 51% at 10 years), followed by those with oligoastrocytoma (70% at 5 years, 49% at 10 years) and fibrillary astrocytoma (65% at 5 years, 31% at 10 years). Survival of patients with gemistocytic astrocytoma was poor, with survival rates of 16% at 5 years and 0% at 10 years [38].
Elsewhere in Europe, data from cancer registries are not specific for DLGG and vary with periods of time and regions. For example, the Danish registry, one of the oldest registries, compared the overall survival of patients with oligodendroglial tumors (oligodendrogliomas and anaplastic oligodendrogliomas) during the periods 1943–1977 and 1978–2002. The median survival increased from 1.4 years (95% confidence interval [CI], 1.0–1.6) to 3.4 years (95% CI, 2.6–4.2) during the period of study [47]. More recently, EUROCARE group (included data from 39 cancer registries located in different regions of Europe) showed that estimates of 5-year relative survival rates (95% CI) for patients with oligodendrogliomas/anaplastic oligodendrogliomas, alive in 2000–2002, were 74.1 (64.4–81.8)/35.1 (21.2–49.5) in Northern Europe, 65.8 (57.5–73.0)/35.5 (24.4–46.9) in UK and Ireland, 75.5 (61.8–85.2)/29.7 (13.4–48.3) in Central Europe, 47.8 (32.4–62.0)/6.1 (1.3–16.6) in Eastern Europe, 63.8 (51.4–74.1)/33.3 (14.7–53.6) in Southern Europe, and 67.2 (62.5–71.6)/31.5 (25.0–38.3) in all cases [48]. The last data of primary malignant brain tumors from EURCARE (including 58 cancer registries, period 2000–2007) have grouped many glioma types, and it is not possible to separate DLGG. But for the group of “oligodendroglioma, anaplastic oligodendroglioma, oligoastrocytoma and anaplastic oligoastrocytoma”, the 5-years relative survival for the subgroup of 45–54 years old patients and the subgroup of 55–64 years old patients were 51.4 and 31.0% in the period 1999–2001, versus 60.4 and 38.5% in the period 2005–2007 with p = 0.005 and p = 0.02, respectively [49]. In the work by Crocetti et al. [50], data from 76 cancer registries out of the 89 that accepted to participate in the RARECARE project were considered. The estimated 5-year relative survival was 14.5% for astrocytic tumors (42.6% for low grade astrocytomas, 4.9% for high grade astrocytomas, and 17.5% for gliomas NOS), and 54.5% for oligodendroglial tumors (64.9% for low grade and 29.6% high grade). Survival data for gliomas including data for some subtypes of DLGG are available in few other countries (e.g., England, Korea, Netherland, and Sweden [51–54]). Ostrom et al. [39] compared some of them.
US population data show that all DLGGs have survival averaging approximately 6 [54]–7 years [55], although variation in survival is quite large with at least 20% of patients surviving for two decades [55]. The median survival for patients with astrocytoma, mixed glioma, and oligodendroglioma is 5.2, 5.6, and 7.2 years, respectively.
One important question is to know if survival of DLGG is improved with the new medical technology. The answer of this question is not so easy.
Data from the Surveillance, Epidemiology and End Results (SEER) program of the National Cancer Institute suggest that for the majority of low grade glioma patients, overall survival has not significantly improved over the past three decades [56]. Data from other, but smaller works showed a modest improvement at least for tumors with oligodendroglial component [47, 49, 53], and some important clinical studies showed a median survival longer than 10 years (e.g., [30]).
Generally speaking, the length of the mean survival of DLGG, the heterogeneous medico-surgical care, the choice of the best personalized therapeutic, the acquisition of experience of new technologies (awake surgery, second or third surgery eventually, chemotherapies, new modalities of radiotherapy, etc.), needs a follow-up of at least 10–20 years to demonstrate an improvement at population level. Moreover, very few registries collect many prognostic factors as functional status, biology, quality of resection, all therapeutic lines, etc. to compare survival.
2.3.3 Prevalence
Prevalence rates are ideally suited to provide an overall estimate of cancer survivorship and direction for health planning as they reflect the complex relationships between incidence, survival, and population demographics—and hence to provide valuable information to the research and medical community. But prevalence data for PCNSTs are limited and very difficult to obtain. In theory, this would imply that the registration of cases is (and has been) exhaustive for many years (to account for long survivors), and that the histological classification systems have not changed over this long period. In 2001, Davis et al. showed that the prevalence rate for all PCNST was 130.8 per 100,000 with approximately 350,000 individuals estimated to be living with this diagnosis in the United States in 2000. The prevalence rate for primary malignant tumors was 29.5 per 100,000, the prevalence rate for primary benign tumors was 97.5 per 100,000, and 3.8 per 100,000 for primary borderline tumors [57]. The same group published new prevalence data in 2010. On the basis of the sum of non-malignant and averaged malignant estimates, the overall prevalence rate of individuals with a PCNST (as defined by CBTRUS) was estimated to be 209.0 per 100,000 in 2004 and 221.8 per 100,000 in 2010. The female prevalence rate (264.8 per 100,000) was higher than in males (158.7 per 100,000). The average prevalence rate for malignant tumors (42.5 per 100,000) was lower than for non-malignant tumors (166.5 per 100,000) [58]. In Europe, Crocetti et al. [50] published that the estimated prevalence rate for all astrocytic tumors of CNS was 20.4/100,000, and the estimated prevalence rate for oligodendroglial tumors of CNS was 2.7/100,000, with an incidence rate (per 100,000 person-years, age standardized on European population) of 4.4 and 0.4, respectively.
If until now, no specific prevalence data for DLGG are available in large population, we could use a crude approximation. If the average duration of disease and the population of patients are stationary, the prevalence can be estimated by the incidence (I) and the mean duration of disease (D) with the following equation: P ≈ I×D. Given the survival data (see previous and following paragraphs and without taking into account that survival could be improved), a crude approximation of the DLGG mean duration could be about 7–12 years. An estimation of the incidence rate for DLGG was ≈1/100,000 person-years (see above). So according to this crude approximation (and with I ≈ 1/100,000 person-years), the approximate value of the prevalence rate for DLGG would be about 10/100,000, or even more if we consider that the mean survival is longer. It is important to notice that the prevalence rate for DLGG is higher than the prevalence rate for glioblastoma.
On the other hand, as it has been noted by Mandonnet et al. [59], DLGG is a progressive primary brain tumor for which several stages can be discerned (i.e., one of them is a long clinical silent stage). Now, it is possible to have an estimation of the prevalence of silent DLGG by MRI performed in healthy subjects. In the conditions of the experiments, the prevalence of DLGG was between 0.1 and 0.2% of the studied population [41–43].
2.4 Prognostic Factors for DLGG
Due to the lack of class I evidence concerning the impact of available treatments for DLGG, and the difficulties to make large clinical trials on DLGG (limited number of patients and long survival), the knowledge of spontaneous prognostic factors is crucial for analyzing the effects of the different therapeutic strategies performed on different populations. Except for age, sex and race, little is known for these factors in population based-studies: so most of prognostic factors come from clinical studies.
2.4.1 Age, Sex and Race
Age is one of the most important spontaneous prognostic factors for DLGG. Survival of diffuse astrocytomas and oligodendroglioma by age groups (CBTRUS 2015 data, period 1995–2012, [18]) are shown in Table 2.7. In the study by Claus and Black [55] entitled “survival rates and patterns of care for patients diagnosed with supratentorial low-grade gliomas (Data from the SEER Program, 1973–2001)”, improved survival was significantly associated with female gender (hazard ratio [HR], 0.84; 95% CI, 0.74–0.95), younger age, white race (HR, 0.70; 95% CI, 0.54–0.93), histology, and later year of diagnosis. In Europe, Crocetti et al. [50] noted that for glial tumors, the 5-year survival was slightly higher for women (20.7%; 95% CI 19.6–21.9) than for men (18.7%; 95% CI 17.8–19.7). Sant et al. [48] found also slightly better survival for women than men only for malignant PCNSTs. For many cancers, women survive longer than men, and this has been attributed to lower prevalence of comorbidities in women, a better performance status (allowing full application of effective surgical and adjuvant treatments) as well as a better “resistance” to disease [60].
Table 2.7
One-, Two-, Five-, and Ten-year relative survival ratesa,b for diffuse astrocytoma and oligodendroglioma by age groups, SEER 18 registries, 1995–2012c, from 2015 CBTRUS report [18]
Age group | Nd | 1-Yr | 2-Yr | 5-Yr | 10-Yr |
|---|---|---|---|---|---|
Diffuse astrocytomae | |||||
0–19 | 992 | 92.7% | 87.0% | 82.7% | 80.3% |
20–44 | 2349 | 92.4% | 85.2% | 65.9% | 47.2% |
45–54 | 1046 | 74.6% | 60.2% | 42.9% | 31.2% |
55–64 | 933 | 54.7% | 34.4% | 21.0% | 12.8% |
65–74 | 712 | 37.6% | 24.3% | 13.4% | 9.3% |
75+ | 603 | 21.3% | 10.8% | 5.4% | 2.0% |
Oligodendroglioma | |||||
0–19 | 273 | 96.7% | 94.7% | 91.9% | 89.3% |
20–44 | 1832 | 98.0% | 95.4% | 86.0% | 68.6% |
45–54 | 792 | 94.2% | 89.1% | 79.1% | 61.8% |
55–64 | 431 | 87.8% | 78.2% | 65.4% | 48.3% |
65–74 | 176 | 77.3% | 68.4% | 50.6% | 34.4% |
75+ | 98 | 61.0% | 50.6% | 38.4% | 18.4% |
2.4.2 Clinical Status
The clinical and neurological status, before and/or after an oncological treatment, classically influences survival [61, 62]. The presence of a neurological deficit increases with age, tumor extension and mass effect [63]. At time of diagnosis, the existence of epilepsy is inversely linked to the presence of a deficit, and consequently carries a favorable prognostic value when isolated [64–66].
2.4.3 Tumor Location, Size and Growth Rates
DLGGs are commonly located in or close to eloquent areas, i.e. those areas of the brain involved in motor, language, visuospatial and cognitive functions [30, 62, 67–69]. Larger tumors and tumors crossing the midline correlate with a shorter survival [64]. Growth rates are inversely correlated with survival [70–72]. It is important to note that DLGGs show a constant linear growth before malignant transformation. Very slow progression is possible, but these tumors always grow. The average slope is about 4 mm of mean diameter per year before and/or after surgical resection (without adjuvant therapy) [73–75].
2.4.4 Prognostic Scores
In a recursive partitioning analysis, Bauman et al. [76] identified four prognostic groups of patients with statistically different median survivals (MS): (1) [KPS <70 and age >40y, MS: 12 m], (2) [KPS ≥70, age >40y, and enhancement present, MS: 46 m], (3) [KPS <70 and age: 18–40y, or KPS ≥70 and age >40y, no enhancement, MS: 87 m], and (4) [KPS ≥70 and age: 18–40y, MS: 128 m], with the following abbreviations: KPS Karnofsky performance Status, y years, m months.
In 22844 and 22845 EORTC trials, Pignatti et al. [64] showed that age >40 years, astrocytoma histology subtype, largest diameter of the tumor >6 cm, tumor crossing the midline, and presence of neurologic deficit before surgery were unfavorable prognostic factors for survival. The total number of unfavorable factors can be used to determine the prognostic score.
In the University of California at San Francisco LGG prognostic scoring system, patients were assigned a prognostic score based upon the sum of points assigned to the presence of each of the four following factors: (1) location of tumor in presumed eloquent cortex, (2) KPS Score ≤80, (3) age >50 years, and (4) maximum diameter >4 cm [77, 78]. Survival estimates according to this DLGG score were applied in 537 patients and are exposed in Tables 2.8 and 2.9.
Yes/No | |
|---|---|
Age >50 years | 1/0 |
KPS ≤80 | 1/0 |
Eloquent cortex location (presumed) | 1/0 |
Maximum diameter >4 cm | 1/0 |
Score | 0–4 |
Table 2.9
Survival estimates (cumulative overall survival probabilities) according to the UCSF hemispheric diffuse low grade glioma score in the combined construction and validation sets (N = 537), adapted from Chang et al. [78]
DLGG score | 0 year | 2.5 years | 5 years
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |||
|---|---|---|---|---|---|---|