Uveal melanoma is the most common primary intraocular malignancy in adults. The disease overwhelmingly affects white populations. Other risk factors include fair skin, light iris color, ancestry from northern latitudes, and ocular/oculodermal melanocytosis. Historically, enucleation was the definitive treatment of uveal melanoma, but brachytherapy and proton beam irradiation are now the most commonly used treatment methods. However, there are still no effective therapies against metastatic uveal melanoma, which is almost always fatal. Continued advances in understanding of the molecular mechanisms of uveal melanoma will facilitate the identification of prognostic markers and therapeutic targets.
Key Points
- •
Uveal melanoma is the most common primary intraocular malignancy in adults.
- •
Most affected patients are Caucasian.
- •
Most cases can be treated with eye-conserving local irradiation, but enucleation is sometimes necessary.
- •
The pathogenesis of uveal melanoma involves the mitogen-activated protein kinase pathway.
- •
Risk of metastatic disease can be predicted by molecular diagnostic testing.
- •
There is currently no effective treatment of metastatic uveal melanoma.
Introduction
Uveal melanoma is the most common primary intraocular malignancy in adults. The tumors arise from the uveal tract, which is comprised of the iris, ciliary body, and choroid. Although both cutaneous and uveal melanomas share the melanocyte as their common origin, their clinical behavior and underlying molecular mechanisms are significantly different. Substantial advances in the diagnosis and local treatment of uveal melanoma have been made in the past few decades, shifting from enucleation to eye-conserving treatments as the primary treatment modalities, without compromising survival.
Introduction
Uveal melanoma is the most common primary intraocular malignancy in adults. The tumors arise from the uveal tract, which is comprised of the iris, ciliary body, and choroid. Although both cutaneous and uveal melanomas share the melanocyte as their common origin, their clinical behavior and underlying molecular mechanisms are significantly different. Substantial advances in the diagnosis and local treatment of uveal melanoma have been made in the past few decades, shifting from enucleation to eye-conserving treatments as the primary treatment modalities, without compromising survival.
Epidemiology
Incidence
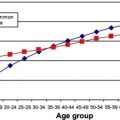
Uveal melanoma is relatively rare. A study based on the National Cancer Data Base of the United States reviewed 84,836 cases of cutaneous and noncutaneous melanomas diagnosed between 1985 and 1994. Ocular melanomas comprised 5.2%, of which 85% were uveal melanomas. In an analysis of the Surveillance, Epidemiology, and End Results (SEER) program of the National Cancer Institute, which covers approximately 28% of the US population, Singh and colleagues reported 4070 cases of uveal melanoma from 1973 to 2008. This rate represented 3.1% of all recorded cases of melanoma. The mean age-adjusted incidence was calculated to be 5.1 per million per year (95% confidence interval [CI], 4.8–5.3). The incidence remained unchanged from 1973 to 2008. Similar rates, or slightly higher, have been reported from Canada, Denmark, Finland, Israel, Norway, and the Netherlands, which have predominantly white populations.
Age
The average age at initial diagnosis of uveal melanoma is approximately 60 years. Unlike other malignancies, uveal melanoma may plateau or decrease in incidence in older patients. Pediatric cases are rare. In several large series of patients with uveal melanoma, only 0.5% to 1.3% were 20 years of age or younger. Compared with older patients, younger patients are more likely to have associated ocular melanocytosis, present with iris melanomas, and lower risk of metastatic disease. Congenital cases have been reported, but seldom occur.
Gender
It is uncertain whether there is a difference in the incidence of uveal melanoma between men and women. Data from the SEER program indicate that the age-adjusted incidence is slightly higher in men: 5.8 per million (95% CI, 5.5–6.2) for men, and 4.4 per million (95% CI, 4.2–4.7) for women. However, a review of 8033 eyes from Wills Eye Institute did not note a difference. The authors also have not observed a gender predisposition at the Massachusetts Eye and Ear Infirmary.
The Collaborative Ocular Melanoma Study (COMS) was a set of multicenter randomized clinical trials in the United States and Canada ( Table 1 ). In the medium-sized melanoma trial, comparing enucleation to brachytherapy, there was no gender imbalance: 1478 men (51%) and 1398 women (49%) were eligible for the study. However, in the large-sized melanoma trial, comparing enucleation with or without preenucleation radiation therapy, there was a slight predominance of men: 731 (56%) men and 567 (44%) women were eligible for the study, but there was no difference in those who were not eligible. An interesting finding from Sweden notes that while there was a decrease in the incidence of uveal melanoma in men between 1960 and 1998, the incidence remained stable in women. In a study of 6673 cases from European registries from 1983 to 1994, there were more men with uveal melanoma than women (incidence rate ratio 1.22, 95% CI, 1.16–1.28).
| Size Category | Apical Height (mm) | Longest Basal Diameter (mm) |
|---|---|---|
| Small | 1.0 to <2.5 | 5.0 to 16.0 |
| Medium | 2.5 to 10.0 | ≤16.0 |
| Large | ≥2.0 | >16.0 |
| >10.0 | any |
a Criteria since November 1990. Please see references for peripapillary tumor criteria.
Race and Ethnicity
Uveal melanoma predominantly affects the white population. In the recent SEER study, 97.8% of affected patients were white, with a white:black incidence ratio of 196:1. In a smaller analysis of the SEER database from 1992 to 2000, Hu and colleagues reported that of 1352 uveal melanomas, the annual age-adjusted incidence per million was 0.31 in blacks, 0.38 in Asian and Pacific Islanders, 1.67 in Hispanics, and 6.02 in non-Hispanic whites. The relative risks compared with black individuals were 1.2 for Asian and Pacific Islanders, 5.4 for Hispanics, and 19.2 for non-Hispanic whites. At Wills Eye Institute, of 2586 patients with posterior uveal melanoma from 1974 to 1987, only 10 (0.39%) were black. A study from the Armed Forces Institute of Pathology reported 39 black individuals out of 3876 (1.0%) with ciliary body and choroidal melanomas before 1975. The investigators noted that compared with a control group of white subjects, black subjects were more likely to have secondary glaucoma and inflammation before enucleation.
Uveal melanoma is rare in Asian populations but may occur at an earlier age. A review of 65 cases of uveal melanoma from Shanghai between 1956 and 1979 noted that 20% occurred in patients between 19 and 30 years of age. Most cases occurred in the fifth decade, but only 5 occurred in the sixth decade, which is the highest risk age group in white populations. A high number of epithelioid tumors were noted, which often portends a poorer prognosis.
Among the white population, ancestry from northern latitudes appears to be a risk factor for developing uveal melanoma. Our group investigated 197 white individuals with uveal melanoma with matched population controls and found that compared with subjects with parents of both Southern European or Mediterranean ancestry (Greece, Italy, Portugal, Spain, Yugoslavia, or Middle East), the adjusted relative risks were 2 (95% CI, 0.96–4.2) for those of central European decent (other European countries and Russia), 2.4 (95% CI, 1.02–5.5) for those of mixed ancestry, 2.4 (95% CI, 1.1–5.1) for those of British decent, and 6.5 (95% CI, 1.9–22.4) for those of northern European decent (Finland and Scandinavian and Baltic countries, including Lithuania, Latvia, and Estonia). Similar results were found in the European Cancer Registry-based study on survival and care of cancer patients (EUROCARE) study that examined 67 cancer registries from 22 European countries. Five thousand five hundred sixty-six cases of uveal melanoma were reported from 1983 to 1994, and standardized incidence rates increased from southern to northern registries, with a minimum of less than 2 per million in Spain and southern Italy, up to greater than 8 per million in Norway and Denmark.
In addition to ethnicity, intrinsic host factors that appear to predispose white individuals to uveal melanoma include light eye color, light skin color, and the inability to tan. A meta-analysis of 10 case-control studies of white populations found that the blue or gray eye color (odds ratio [OR] 1.75 [95% CI, 1.31–2.34]), fair skin color (OR 1.80 [95% CI, 1.31–2.47]), and a propensity to sunburn (OR 1.64 [95% CI, 1.29–2.09]) were statistically significant risk factors.
Choroidal Nevi
Cutaneous nevi are a well-known risk factor for developing cutaneous melanoma. Uveal melanomas are thought to arise from choroidal nevi as well, but the evidence is not as robust. Approximately 3% of individuals older than 30 years of age have nevi in the posterior half of the choroid, and it has been estimated that 1 out of every 4800 choroidal nevi may transform into a melanoma per year. Similarly, the Blue Mountains Eye Study estimated the rate of malignant transformation to be 1 melanoma per 4300 nevi per year. A systematic literature review noted the prevalence of choroidal nevi to be 4.6% to 7.9% in the white US population and estimated the annual malignant transformation to be 1 in 8845. In clinical practice, benign-appearing choroidal nevi are monitored on regular eye examinations. Fundus photography and ultrasonography are valuable adjuncts to the clinical examination in determining if there has been significant growth.
Ocular/Oculodermal Melanocytosis
Ocular melanocytosis is a diffuse congenital melanosis of the episclera and uvea that is more common in black, Hispanic, and Asian populations. Half of the patients also have ipsilateral dermal melanocytosis (nevus of Ota), and the combination is termed oculodermal melanocytosis. Ocular/oculodermal melanocytosis affects approximately 0.04% of the general white population, whereas about 1.4% of patients with uveal melanoma have ocular melanosis, indicating that ocular melanosis is approximately 35 times more common in patients with uveal melanoma. One in 400 patients with ocular melanosis is thought to develop uveal melanoma in their lifetimes. The increased number of melanocytes in the uveal tract appears to predispose these patients to uveal melanoma, as evidenced by their propensity to develop the tumors in sectoral areas affected by the melanocytosis.
Other Risk Factors
Development of uveal melanoma is usually considered a sporadic event. However, approximately 90 cases of familial uveal melanoma have been reported in the literature, and recently, germline BAP1 (BRCA1 [breast cancer 1, early onset]-associated protein 1) mutations have been shown to predispose to uveal melanoma, cutaneous melanocytic tumors, and other malignancies. Pregnancy, ovarian cancer, and history of malignancy in women have been implicated as well but without substantial evidence. Some reports indicate that sun exposure may increase the risk of uveal melanoma, but study results are conflicting. Dietary factors and smoking have not been found to be contributory risk factors.
Pathogenesis
Advances in understanding the molecular mechanisms of uveal melanoma have been made in recent years. Cutaneous and uveal melanomas differ in their pathogenesis, despite sharing the melanocyte as their common origin. For example, BRAF (v-raf murine sarcoma viral oncogene homolog B1) mutations are found in up to 62% of cutaneous melanomas but rarely implicated in uveal melanoma. The mitogen-activated protein kinase (MAPK) pathway, of which BRAF is a key component, has been shown to be activated in 86% of primary uveal melanoma specimens. Therefore, cutaneous and uveal melanomas appear to activate the MAPK pathway, but through different mechanisms.
The upstream modulator of the MAPK pathway in uveal melanoma was unknown until recently. Van Raamsdonk and colleagues reported that guanine nucleotide binding protein q polypeptide (GNAQ) or GNA11 mutations occurred in 83% of the 186 uveal melanomas that they analyzed. Both GNAQ and GNA11 encode the alpha subunit of G proteins, which are a family of heterotrimeric proteins bound to cell membrane receptors. These proteins modulate the signaling pathway that lead to activation of downstream components including Raf, MEK, and ERK. GNAQ/11 mutations are found in blue nevi and are not associated with survival in uveal melanoma and therefore, represent early or perhaps initiating mutations that require further genetic aberrations for malignant transformation.
Specific genetic alterations have been associated with increased risk of metastasis of uveal melanoma. Monosomy 3 occurs in approximately half of uveal melanomas and has been strongly correlated with poor histopathologic factors and metastasis-related death. Recently, Harbour and colleagues reported that 26 (84%) of 31 metastasizing uveal melanomas contained BAP1 mutations. BAP1 is located on chromosome 3, and biallelic inactivation of this gene likely accounts for the noted association between monosomy 3 and risk of metastasis.
Clinical presentation and diagnosis
Uveal melanomas are either detected on routine eye examinations, or when patients present with visual symptoms. Common symptoms include blurry vision, photopsias (flashing lights), and visual-field defects. Loss of vision is usually caused by tumor involvement of the macula or by exudative retinal detachments.
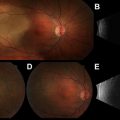
A dilated fundus examination by an ophthalmologist with experience in ocular oncology is the most important factor in accurately diagnosing uveal melanoma. Classically, melanomas appear as pigmented, dome or collar-button shaped masses ( Fig. 1 ). The collar-button configuration occurs when the tumor breaks through Bruch membrane, which is the basement membrane of the retinal pigment epithelium (RPE) and choriocapillaris. Larger tumors may be associated with dilated episcleral vessels, visible externally.
The color, shape, and number of lesions are important considerations in diagnosis. Jet-black pigmentation is not characteristic of uveal melanoma, and other diagnoses should be considered, such as RPE hyperplasia, RPE hypertrophy, and melanocytoma. Amelanotic uveal melanomas do occur, but choroidal hemangioma, choroidal osteoma, and choroidal metastasis should be included in the differential diagnosis of amelanotic lesions ( Fig. 2 ). Multifocal and bilateral lesions are also more consistent with choroidal metastasis. Hemorrhage, inflammation, and pain are rare but may be seen in large tumors. An atypical presentation is the “diffuse” melanoma, which by definition, is less than 5 mm thick and encompasses more than a quarter of the uvea. Other choroidal tumors that can appear similar to uveal melanoma are listed in Box 1 .
- •
Metastatic carcinoma
- •
Choroidal hemangioma
- •
Combined hamartoma of the retina and RPE
- •
Choroidal osteoma
- •
Intraocular or metastatic lymphoma
- •
Hemangiopericytoma
- •
Leiomyoma
- •
Neurilemmoma
- •
Adenoma of the pigmented ciliary epithelium
- •
Reactive lymphoid hyperplasia
Abbreviation: RPE, retinal pigment epithelium.
Small lesions are difficult to diagnose definitively. Indeterminate lesions are monitored for growth. Signs suggestive of malignancy include tumor thickness greater than 2.0 mm, posterior tumor margin abutting the optic disc, visual symptoms, orange pigment (representing lipofuscin deposits at the level of the RPE), and subretinal fluid.
Ultrasonography is the most important ancillary imaging modality in diagnosing and monitoring uveal melanoma. B-scan ultrasonography is useful for characterizing the tumor and for obtaining tumor dimensions. Typical findings in larger melanomas include choroidal excavation and orbital shadowing. High-resolution ultrasound (ultrasound biomicroscopy) allows excellent visualization of iris and ciliary body tumors, which are difficult to fully appreciate on the clinical examination. A-scan ultrasonography classically reveals low to medium internal echogenicity in uveal melanomas, allowing differentiation from tumors with typically high internal reflectivity, such as choroidal hemangiomas.
Fluorescein angiography can also aid in the diagnosis, especially if other lesions that mimic uveal melanomas may be under consideration. Larger melanomas may exhibit an intrinsic tumor circulation called a “double circulation.” Although most cases of uveal melanoma can be accurately diagnosed noninvasively, fine-needle aspiration for cytological studies may be warranted for diagnostic dilemmas, especially if it alters subsequent management. Posterior tumors are accessed via transvitreal approaches and anterior tumors via direct transscleral approaches. Tumor seeding of the biopsy tract does not occur if properly performed by experienced ocular oncologists and retina surgeons.
Treatment
The main treatment options for uveal melanoma are radiation therapy or enucleation. Local resection has been performed for selected tumors. Currently, most patients undergo radiation treatment, and enucleation is reserved for large tumors in which radiation would have high ocular morbidity with little chance of globe or vision retention. The COMS confirmed equivalent survival rates in patients treated with enucleation versus I-125 brachytherapy. The trial enrolled 1317 patients with medium-sized tumors, and the 5-year survival rate for enucleation was 81% (95% CI, 77%–84%), and 82% (95% CI, 79%–84%) in the brachytherapy arm. This rate was not a statistically significant difference. Further analysis indicated that 12.5% (95% CI, 10.0–15.6%) of eyes initially treated with brachytherapy required enucleation in the first 5 years. Visual acuity loss to 20/200 or worse occurred in 43% (95% CI, 38%–48%) of eyes in the first 3 years. The 12-year follow-up data confirmed that enucleation does not confer a survival advantage.
Brachytherapy
Radiation therapy has become the most commonly used treatment modality for choroidal melanoma since the COMS trials. Brachytherapy is the most frequently treatment because of its wide availability. Cobalt-60 was initially used in the 1970s and 1980s, but the high-energy gamma emission did not allow adequate shielding of surrounding tissue and operating room staff. The lower-energy iodine-125 plaque was subsequently developed and has largely replaced Cobalt-60 as the isotype of choice in the United States. Other isotopes include palladium-103 and ruthenium-106, but clinical experience is limited.
Ophthalmologists determine the exact location, size, and shape of the melanoma, and physicists subsequently design patient-specific plaques based on such parameters. The plaques are fixated onto the globe in the operating room under intravenous sedation and retrobulbar block. The conjunctiva is first dissected 360°, and the rectus muscles are isolated with sutures for traction. Transillumination is then performed to visualize the tumor borders, which are marked. The plaques are oversized so that they are slightly larger than the actual tumor. A plastic template is first sutured to optimize positioning. The template is then removed, and the radioactive plaque is sutured onto the sclera. Rectus muscles may need to be temporarily disinserted for proper plaque placement. The eye is covered by a lead shield throughout the treatment period, and contact with others are limited to prevent radiation dissemination. The plaque is subsequently removed in the operating room after several days of treatment.
Proton Beam Irradiation
The second major radiation treatment modality for uveal melanoma is charged particle irradiation using protons and helium. Unlike conventional external beam radiation therapy, charged particles allow tightly localized radiation. Helium ion irradiation has been largely replaced by proton beam irradiation because of its cost. Protons and helium ions are charged particles with a property called the Bragg peak, which allows a uniform radiation dose at desired depths, with a sudden drop off of radiation, thus theoretically limiting irradiation of surrounding tissues. Large tumors and posterior tumors that cannot be accessed by conventional plaque therapy are often referred to proton centers. The gamma knife is an alternative stereotactic option to proton beam irradiation, but there is limited data, and the dose distributions are less favorable compared with proton therapy.
Proton treatment requires tumor localization surgery in which tantalum rings are sutured on the sclera to outline the tumor. The relative position of the rings as seen on radiograph are used in a treatment planning program, which creates a 3-dimensional model of the eye and the tumor to help determine the best gaze direction and specify beam parameters. Standard treatment of uveal melanoma uses 70 cobalt gray equivalents delivered in 5 fractions over 5 to 7 days. Individualized facemasks and bite blocks are mounted onto the treatment frame for immobilization, and the patient is asked to gaze at a fixation light. Fluoroscopy is used to confirm positioning, and eye movement is monitored by a closed-circuit television system.
Most tumors show regression by 6 months and continue to regress thereafter. The risk factors for poor visual outcome include proximity to the macula and optic nerve, tumor height and diameter, baseline visual acuity, retinal detachment, and history of diabetes. For example, for patients with tumors less than 5 mm in height and located more than 2 disc diameters away from the optic nerve and fovea, approximately 75% of patients retain vision of 20/200 or better at 10 years, whereas the rate is only 3% for those with tall tumors close to both the optic disc and fovea.
Local recurrence is observed in 2% to 5% of eyes treated with charged particle irradiation. At the Massachusetts Eye and Ear Infirmary, of 2069 tumors treated with proton beam irradiation, 2.2% showed growth between 5 months and 11 years after treatment. This recurrence rate is favorable compared with rates reported with brachytherapy. Main risk factors for tumor recurrence are larger tumor diameter and tumors that involve the ciliary body. Local recurrences are typically treated with repeat proton irradiation or with enucleation. However, recurrences are associated with an increased risk of death from metastatic disease.
Complications of Radiation Treatment
Radiation retinopathy is a slowly progressive disease caused by radiation-induced endothelial damage and capillary occlusion, which results in retinal hemorrhage, macular edema, vascular sheathing, microaneurysms, retinal exudation, telangiectasias, retinal pigment epithelial atrophy, and cotton wool spots. Radiation optic neuropathy is characterized by optic disc hemorrhage, disc pallor, and/or disc edema and was present in 27.4% of subjects in the COMS trial at 5 years. In a review of 1300 patients treated with brachytherapy, 42% (95% CI, 38%–45%) developed radiation retinopathy at 5 years. Major risk factors for developing retinopathy were high radiation dose and proximity of the tumor to the fovea and/or optic disc. In patients treated with proton beam irradiation, the incidence of radiation retinopathy and optic neuropathy peaks at 2 to 3 years after treatment, and at 10 years, approximately 50% patients have retinopathy and 25% have optic neuropathy. Diabetes mellitus has been shown to be a significant risk factor for developing radiation retinopathy.
The main treatment of radiation retinopathy is panretinal photocoagulation for proliferative disease (in which there is retinal neovascularization). Emerging therapeutic modalities for macular edema in this setting include intravitreal or periocular steroids, intravitreal anti-vascular endothelial growth factor (VEGF) agents, and photodynamic therapy. There is no proven treatment of radiation optic neuropathy; mixed results have been reported for corticosteroids, hyperbaric oxygen, and anticoagulation. Intravitreal anti-VEGF treatment has been proposed as a possible treatment option, but the evidence is lacking. The authors’ study investigating the natural history of radiation optic neuropathy in eyes with parapapillary melanomas treated with proton beam irradiation showed that of 93 patients, 42% retained visual acuity at counting fingers or better at 5 years, and 31% had spontaneous visual improvement of 3 or more lines, indicating that the natural history of radiation optic neuropathy after proton irradiation may be better than generally assumed.
Metastatic Disease
Overall, approximately half of patients with posterior uveal melanoma will die from the disease. Uveal melanoma metastasizes hematogenously because there are no lymphatics in the uveal tract. Unlike cutaneous melanoma, the liver is the most common site for metastasis, accounting for 90% of cases. The factors associated with metastasis include tumor size and location (see Table 1 ), histologic grade, and genetic composition. Ciliary body-involving tumors are more likely to metastasize, whereas iris melanomas are significantly more indolent, and epithelioid tumors are more aggressive than spindle tumors. Two TNM staging systems exist (one for all uveal melanomas and one for ciliary body/choroidal melanomas), based on size and location (ciliary body vs choroidal), and presence of extrascleral extention. However, TNM staging has limited clinical application.
Risk of metastasis can be most accurately predicted by molecular profiling of fine-needle aspiration biopsy samples of the ocular tumor before irradiation. Cytogenetic analysis for monosomy 3 status and expression profiling are the 2 most commonly used methods for risk profiling. A 15 gene expression profiling assay has been shown to have better accuracy in predicting risk of metastasis than monosomy 3 status ( Fig. 3 ). Expression profiling defines 2 classes of uveal melanoma: class 1: tumors which have extremely low rates of metastasis and class 2: tumors in which approximately 50% of patients have metastatic disease at 3 years after diagnosis.