and Karl Reinhard Aigner3
(1)
Department of Surgery, The University of Sydney, Mosman, NSW, Australia
(2)
The Royal Prince Alfred and Sydney Hospitals, Mosman, NSW, Australia
(3)
Department of Surgical Oncology, Medias Clinic Surgical Oncology, Burghausen, Germany
In this chapter, you will learn about:
Comparative cancer incidence
People most at risk
Viral and other infection associations
Heredity and genetic factors
Age
Predisposing and pre-malignant risk factors
Gender
Diet
Race
Geographic associations
Environment
Occupation and cancer
Habits and lifestyle
Possible psychological factors
Cancer registries
2.1 Comparative Cancer Incidence
The incidence of cancers is very different in different countries (see Appendix). The most obvious differences are between developed and developing countries. Cancers of the lung, prostate, breast, colorectum and pancreas are all much more common in developed countries, and cancers of the oesophagus and liver are much more common in developing countries. Other cancers that are at least twice as common in developed countries are cancers of the skin, uterus, ovary, bladder, kidney, testis, brain, lymphomas, leukaemias and multiple myeloma. Nasopharyngeal cancers are more than twice as common in developing countries.
The incidence of cancers is very different in different countries (see Appendix). The most obvious differences are between developed and developing countries.
Table 2.1 shows the year 2001 incidence of the most common internal cancers in the United States and the incidence of death rates from these cancers.
Table 2.1
The year 2001 incidence of the most common internal cancers in the United States and the incidence of death rates from these cancers
Cancer | Incidence | Deaths |
|---|---|---|
Prostate | 198,100 | 31,500 |
Breast | 193,700 | 40,600 |
Lung | 169,500 | 157,400 |
Colon and rectum | 135,400 | 56,700 |
2.2 People Most at Risk
Although the risk of developing cancer is much lower in young people, cancer can affect people of any age, race or occupation in any part of the world. People who have been cured of one cancer often ask about the risk of developing a second cancer. Whilst it is true that some people have an increased predisposition towards developing cancer, in most cases, people who have already had one cancer cured have only a slightly greater risk of developing a second cancer than do people who have never had a cancer. For example, a woman who has been cured of breast cancer has an increased risk of developing cancer in the other breast and a somewhat increased risk of developing cancer of the uterus or ovary, but the majority of these people never develop any other serious cancer. Again, people who have been treated and apparently cured of one bowel cancer do have an increased risk of developing a second cancer elsewhere in the bowel, but most do not. It is also true that people who have been cured of one type of cancer have a slightly increased risk of developing a second cancer, not only of the same system but of another system of the body, but the increased risk is small. However, the risk of developing a second cancer is increased if they continue to indulge in an obviously cancer-causing habit such as cigarette smoking or if they inherit a mutation in a tumour suppressor or proto-oncogene. There is also an increased risk of developing a leukaemia in some people 20 years or so after treatment of another cancer with a prolonged chemotherapy program.
2.3 Viral and Other Infection Associations
In the normal course of events, cancer cannot be passed directly from one individual to another.
In the normal course of events, cancer cannot be passed directly from one individual to another. The present day worldwide scourge of acquired immune deficiency syndrome (AIDS) is caused by a virus infection that can predispose to cancer, but AIDS is not itself a cancer. In this disease, the sufferer’s natural immune defences against infection and cancer are damaged, resulting in a higher incidence of cancer developing in affected people. Several cancers are now commonly associated with HIV infection or AIDS. These include a sarcoma of soft tissues called Kaposi’s sarcoma, lymphomas of the central nervous system, non-Hodgkin lymphoma and cancer of the cervix. Each of these is described and discussed in Part 3 of this book.
Similarly, liver cancer is not infectious, but a common precursor of liver cancer is the chronic inflammatory changes in the liver due to hepatitis B or hepatitis C infection. These hepatitis infections do spread easily from person to person, mainly from food or intimate contact. In the case of hepatitis C, blood transfusion or sharing of intravenous needles is also a common method of spread. Liver cancer therefore develops more commonly in people who have been infected. However, in many cases, liver cancer doesn’t develop until at least 20 years after infection. This is known as “the latency period” and is consistent with the clonal origin of cancers, and 5–11 other genetic alterations are required for cancer to develop.
The human papillomavirus is sometimes responsible for papillomas or squamous cell carcinomas of the skin or genitals of either sex. It is often transmitted during sexual intercourse and is particularly associated with cancer of the cervix. The latency period for the development of cervical cancer from human papillomavirus is 5–30 years.
Viruses can act by modulating the function of proto-oncogenes and tumour suppressors. These original findings provided initial molecular insights into the genetic basis of cancer.
2.4 Heredity and Genetic Factors
Molecular biology and studies of association of genes with cancer is one of the most stimulating, challenging and exciting fields of cancer research.
At this stage, information about three genetic concepts is important. These still-evolving concepts are about cancer proto-oncogenes, tumour suppressor genes and cell cycle regulation genes. It is certain that these concepts will be modified, changed or added to fairly rapidly as more information is gathered in this relatively new but exciting area of study being carried out in many laboratories in many parts of the world. It is not known whether all the different carcinogenic factors already described activate “biological triggers” in cells. Whatever the mechanism or combination of mechanisms, it seems that cancers are ultimately caused by changes in tumour suppressor genes or proto-oncogenes that convert them into oncogenes, i.e. genes that can cause cancer.
About 10 % of breast cancers result from breast tissue changes due to one of several specific genes that have been inherited from a parent.
2.4.1 Tumour Suppressors, Proto-Oncogenes and Cancer Oncogenes
The many functions of our body cells are controlled by genes. Genes are coded in the DNA that makes up the chromosomes or genetic library of our cells. Like the genes that determine features like eye colour and blood group, we inherit these controlling genes from our parents. There is a link between genetic make-up and some cancers.
Different genes are associated with different cancers; for example, the BRCA1 gene is often associated with either breast cancer or ovarian cancer. The BRCA2 gene can be associated with either breast cancer or pancreatic cancer. Recent studies have also shown a link between the BRCA2 gene and prostate cancer, particularly prostate cancer in younger men.
The p53 gene is the gene most commonly associated with a broad spectrum of cancers. This gene is responsible for coordinating the cellular response to DNA damage, be it a transient growth arrest to allow the cell to repair the DNA damage or to instruct the cell to commit suicide via apoptosis if the damage was too great. p53 protein is a transcription factor that switches on expression of genes that regulate the cell cycle and cause growth arrest and apoptosis. Accordingly, it has been called the guardian of the genome because of its role in indirectly maintaining the coding integrity of the genetic blueprint. Approximately half of all cancers carry an abnormal (mutated) p53 gene and have lost the other normal copy. Every normal cell has two copies of every gene (except some genes of the X and Y chromosome in males). The mutation of one p53 gene leaves the other one potentially active and able to regulate cell growth and apoptosis. However, the mutant p53 gene makes a protein that inactivates the normal p53 by binding to the normal protein and inactivating the normal p53 protein. Since the protective role of normal p53 protein is now overcome, the genetic material is unstable and the remaining normal p53 gene is deleted from its position on chromosome 17. The mutation of one gene followed by the loss of the remaining normal gene is a common feature of tumour suppressor genes. The mutant p53 gene by indirectly promoting cancer behaves as an oncogene, so the p53 gene can behave as both a tumour suppressor and an oncogene depending on whether or not it is mutated.
BRCA1 and BRCA2 are also tumour suppressor genes and like the p53 gene make a protein that has a role in switching on gene expression and is involved with DNA repair and regulation of the cell cycle.
All tumour suppressor genes play a modulating or inhibitory role in cell growth and differentiation. Factors that impair or damage these genes can therefore be carcinogenic.
Cells also contain special genes called proto-oncogenes. These proto-oncogenes are responsible for programmed growth in development or repair. They play a major role in coordinating our growth from a single fertilised egg cell into an adult with 1013 cells. When development or tissue repair is complete, the cell growth is switched off. Cancer-causing agents or spontaneous genetic mutation changes the proto-oncogenes into potentially cancer-causing oncogenes, as they promote growth where and when they should not. Spontaneous genetic mutation increases as we get older as our DNA repair processes become less efficient. When an oncogene is active in a cell, the cell doesn’t require growth signals to grow so that the “switched-on” mechanism of growth and repair continues instead of being “switched off” as it should be, and the cells that are produced do not later undergo apoptosis (self-destruction) when they are not wanted. Unlike tumour suppressors, only one genetic change is associated with these genes becoming oncogenes, as a mutation leads to the activation of the gene function in the absence of the appropriate cell growth signal. Proto-oncogenes or oncogenes are genes that encode proteins involved in all aspects of the cell signalling pathway that promote the social behaviour of cells and their growth.
Cancer-causing oncogenes or defective tumour suppressors may be inherited or result from cancer-causing agents changing proto-oncogenes into oncogenes or from accidental genetic mutation caused by errors in copying the genetic material in dividing cells, by genetic damaging agents within the cell (e.g. oxygen free radicals) or by external agents such as UV irradiation of sunlight. Occasionally, these errors allow cells to divide without correctly partitioning the genetic complement of cells equally between the daughter cells, such that cells now have multiple copies of the genetic material contributed by both parents and become polyploid (have more DNA content per cell). This is a typical feature of cancer cells at times of rapid cell growth in some tissues or after the many years of cell division during the course of a normal life.
In colon cancer, molecular oncologists have identified the sequential genetic changes in specific oncogenes and tumour suppressors that lead a normal cell into becoming a cancer cell.
From breast cancer studies, it has been shown that about 10 % of breast cancers result from breast tissue changes due to one of several specific genes that have been inherited from a parent. Most of the remaining 90 % are probably the result of an accidental genetic mutation after constant and repeated changes in breast tissue over many years during cyclical hormonal stimulation.
2.4.2 Tumour Suppressor Genes
As opposed to proto-oncogenes or cancer oncogenes, these inherited genes, tumour suppressor genes, play a modulating or inhibitory role in cell growth and differentiation. Factors that impair or damage these genes can therefore be carcinogenic.
2.4.3 Cell Cycle Regulatory Genes
In laboratory culture, cancer cells can grow and divide every 24 h; however, in patients, regulatory processes restrict cell doubling times to between 5 and 700 days, depending on the cell type and tumour stage. The control of cell growth and division has been well studied. The cell growth cycle consists of five stages. Stage 1 starts at the end of mitosis (M) and ends at the point the genetic material is copied and is called the G 1 phase (G = gap). This copying of the genetic material defines the next phase, known as the S phase (S = synthesis). The next stage starts at the end of DNA synthesis and ends with the cell starting mitotic division into daughter cells. The gap between the S phase and mitosis (M) is known as G 2. Cells not in cycle or undergoing differentiation are said to be in G 0, as they are no longer in cycle.
The cycle cannot proceed without a series of successive events occurring. If an event fails, then the cell arrests at defined checkpoints to allow adjustments to be made. The most notable checkpoints occur at the G 1 − S and G 2 − M transition points. The G 1 − S checkpoint allows the cell to repair any DNA damage before it is copied in the S-phase, to prevent mutations becoming fixed in the genetic material. The second, G 2 − M checkpoint allows the cell to ensure that the chromosomes are arranged correctly prior to segregation to the daughter cells (Fig. 2.1).
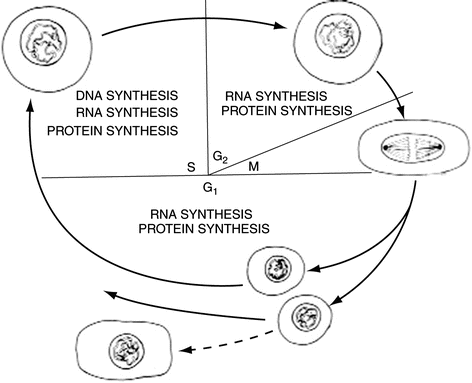
Fig. 2.1
Diagram illustrating the cycle of cell division and the sites of controlling genetic and molecular biological activity. Normal cells and cancer cells divide in a similar way but normal cells stop dividing when sufficient new cells have been made. Cancer cells keep dividing uncontrollably. The new abnormal and unwanted cells become invasive and dangerous, damaging other cells and tissues. Because they are constantly dividing, they are therefore more constantly exposed to anti-cancer drugs that predominantly affect dividing cells when they are in one or more of the stages of cell division (see Sect. 8.3.4)
2.5 Molecular Biological Changes in Controlling Cell Division
The cell cycle control system is based on two partners, cyclins and cyclin-dependent kinases. Cyclins are regulatory proteins expressed at specific stages of the cell cycle that interact with specific cyclin-dependent kinases. The partnerships between the cyclins and the kinases they specifically activate ensure that environmental factors and the cell’s readiness to divide directly influence progress through the cell cycle. The basis of the G 1 − S checkpoint is well understood. Cells can enter S phase only if a certain protein (called the retinoblastoma protein after the eye cancer it is associated with) is phosphorylated by a specific cyclin-activated kinase. The multiply phosphorylated RB protein then releases specific transcription factors, previously bound in an inactive form to itself, to allow them to switch on genes involved in DNA synthesis.
In times of genetic damage, p53 tumour suppressor protein blocks the G 1 − S transition by switching on expression of a protein, p21, that inhibits this kinase to block DNA synthesis and stops the cells entering the S phase.
Since cancer cells exhibit unregulated growth, it is not surprising that they have been shown to have genetic changes in cyclins, cyclin-dependent kinases, p53 and RB protein, to allow the cancer cells to proliferate. The nature of the genetic changes varies from cancer to cancer, which is why it has been difficult to identify the mechanisms of how cells become cancer cells.
More recently discovered are genes that inhibit or stimulate cell reproduction according to need. Impairment of these genes and their active roles in cell reproduction management can also lead to cancer.
2.5.1 Inherited Cancer Genes: Inherited and Familial Cancers
From an immediate practical and clinical point of view, in some relatively uncommon cancers, there is a strong hereditary factor, whilst in other cancers, there is a less obvious hereditary factor; but for most, there is no evidence of a hereditary factor at all. Among the most obvious cancers with a strong hereditary factor as a condition called familial polyposis coli, caused by transmission of a responsible oncogene, half the children of an affected parent are likely to develop the condition of multiple polyps in the large bowel. All those affected who do develop these particular polyps will eventually develop cancer in the bowel, usually by the age of 40.
With a rare but inherited familial condition called xeroderma pigmentosa, there is a high incidence of development of skin cancer.
Another rare but most often inherited condition is called the Li-Fraumeni syndrome. This syndrome is caused by germ-line mutations in the p53 gene. This condition has provided a model to understand further the pathogenesis of genetic cancers. Patients with this syndrome are defective in one allele of the p53 (“the guardian of the genome”) or were born with one non-functional allele of it. Therefore, if the other p53 allele is affected during their life, cancer will develop.
Among the more common cancers with an increased familial incidence are cancers of the breast, stomach and bowel. Although the increased risk for relatives of sufferers is not great in most families, occasionally it may be considerable. For example, there have been rare reports of families in which about half the female blood relatives have developed breast cancer. What is responsible for this apparent increased risk in a few families was largely unknown until recent discoveries of inherited genes called BRCA1 and BRCA2 (the names are derived from BR of breast and CA of cancer). The BRCA1 gene is on chromosome 17 and the BRCA2 gene is on chromosome 13. These genes behave as oncogenes on breast cells (see hereditary and genetic factors). Families with a high incidence of breast cancer may carry one or both of these genes; alternatively, it may be simply that members of these families have similar lifestyle habits and have been affected by similar environmental factors.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


