Fig. 12.1
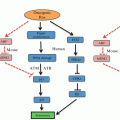
ER stress and unfolded protein response signaling pathway. Upon accumulation of unfolded or misfolded proteins in the ER, three major ER stress sensors, IRE1, ATF6, and PERK are activated following their dissociation from the ER chaperone grp78. Each ER stress sensor is capable of inducing the expression of several major ER chaperones and enhancing protein folding machinery
All three ER stress response sensors are embedded in the ER membrane where they are normally bound by the ER chaperone grp78 (alias BiP) (Kimata et al. 2004; Ma et al. 2002; Sommer and Jarosch 2002). This binding inhibits the activity of each sensor. Grp78 releases the sensors in response to mounting ER stress as its chaperone functions are required (Lee 2005). However, this is not a uniform method of control over the three combined sensors; different cellular conditions result in differing patterns of sensor activation. For example, during B cell differentiation only two sensors, IRE1 and ATF6 are activated while the PERK is not (Ma et al. 2010). Using a B cell line capable of induction of all three ER stress sensors and capable of differentiation into plasma cells, Ma et al. demonstrated that IRE1 is activated quickly upon exposure to differentiation-inducing LPS treatment, with ATF6 activation following secondarily. In contrast, PERK activation could not be elicited from these cells upon differentiation, even when treated with the ER stressor thapsigargin, although this treatment could stimulate PERK activity before differentiation (Ma et al. 2010).
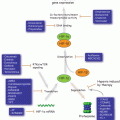
Crosstalk between the sensor systems provides additional control over the cellular response. For example, one effect of IRE1 activation is the transcription of a PERK inhibitor named p58ipk (Iwakoshi et al. 2003; Ma et al. 2010). In addition, ATF6 and PERK appear to converge on signaling through the transcription factor CHOP (Okada et al. 2002). Thus, both re-enforcement and antagonism exist between the sensors, allowing a highly tunable response based on cellular needs. Upon the mild ER stress, ER stress sensors activate the adaptive UPR. However, if UPR stress persists, some of the same UPR sensors activate an ER stress-induced programmed cell death (ER-PCD) (Fig. 12.2).
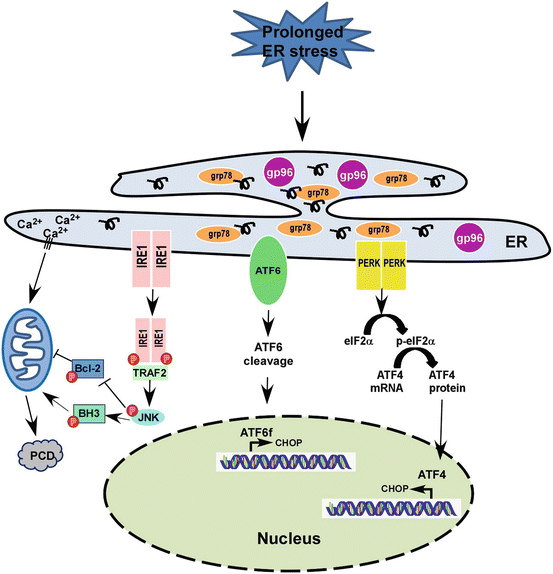
Fig. 12.2
Prolonged ER stress and programmed cell death signaling pathway. Upon persistent ER stress, tipping the system towards programmed cell death (PCD). ATF4 and ATF6 coordinate to induce transcription of CHOP, a pro-apoptosis transcription factor, the ER stress response moves from adaptive to destructive. IRE1 is phosphorylated and binds to TRAF2, then further active JNK-BH3 pathway and induces PCD
Due to the baseline ER stress present in untransformed plasma cells, myeloma is a particularly complex disease in which to examine ER stress. In this chapter, we discuss the roles of ER stress sensors in normal plasma cell differentiation as well as MM pathogenesis. We also summarize the strategies of targeting UPR pathways and HSPs for potential therapeutic benefit against multiple myeloma.
12.2 The Role of ER Stress in Multiple Myeloma: Molecular Mechanisms
Multiple myeloma (MM) is an incurable plasma cell neoplasm. As multiple myeloma cells actively synthesize and secrete immunoglobulin, they are prone to ER stress and require UPR for survival. In this section, we provide brief overviews of the UPR signaling in myeloma disease.
12.2.1 IRE1 and Multiple Myeloma
12.2.1.1 Overview
IRE1 is a bifunctional transmembrane kinase and endoribonuclease. It was first identified in yeast, called Ire1p, which is implicated in unfolded protein response (UPR) (Cox et al. 1993). Upon activation of the UPR, Ire1p oligomerizes, phosphorylates and initiates splicing of HAC1 (Shamu and Walter 1996; Sidrauski and Walter 1997). IRE1 is conserved in all eukaryotic cells. In mammalian cells, there are two forms of IRE1, IRE1α and IRE1β. Most cells and tissues express IRE1α, while only intestinal epithelial cells express IRE1β. IRE1α and IRE1β have similar cleavage specificities (Patil and Walter 2001; Tirasophon et al. 1998; Wang et al. 1998). Previous studies have demonstrated that X box binding protein 1 (XBP1) mRNA is a substrate for the endoribonuclease activity of IRE1. Upon activation of the UPR, the IRE1 RNase activity initiates and removes a 26 nucleotide intron from XBP1 mRNA (Calfon et al. 2002; Lee et al. 2002; Yoshida et al. 2001). This splicing form of XBP1, denoted XBP1s, is a transcriptional activator that plays an important role in activation of a variety of UPR target genes, including ERdj4, p58IPK, DnaJ/Hsp40-like genes, EDEM, HEDJ, protein disulfide isomerase-P5 (PDI-P5), and ribosome-associated membrane protein 4 (RAMP4) (Lee et al. 2003).
12.2.1.2 IRE1/XBP1 Pathway Is Essential for Plasma Cell Differentiation
Both IRE1 and XBP1 are critical for plasma cell differentiation. Genetic deletion of XBP1 causes lack of plasma cells, with concomitantly decreased baseline and antigen specific serum level of immunoglobulin (Iwakoshi et al. 2003; Reimold et al. 2001; Shaffer et al. 2004). In addition, IRE1α is required to splice XBP1 for terminal differentiation of mature B cells into antibody-secreting plasma cells as demonstrated by using an IRE1α-deficient chimeric mouse model (Zhang et al. 2005). Furthermore, in IRE1α conditional knockout mice, the serum levels of IgM and IgG1 are reduced by half compared with the control mice. However, the IgM+, IgD+ and B220+ populations are similar between IRE1α conditional knockout mice and control mice. This result suggests that IRE1α is required for efficient plasma cell production of antibodies, and is critical for final B cell differentiation into a plasma cell (Iwawaki et al. 2010). Very interestingly, the role of XBP1 in plasma cell differentiation does not appear to be due to increased Ig synthesis. It is required for optimal B cell receptor signaling as well as migration of plasmablast to the proper niche in the bone marrow (Hu et al. 2009). These studies suggest that the IRE1/XBP1 pathway is required for differentiation and survival of plasma cells.
12.2.1.3 IRE1/XBP1 Pathway in Multiple Myeloma Pathogenesis
In addition to the critical role of UPR in plasma cell differentiation, XBP1s and the downstream ER chaperones are consistently upregulated in myeloma patients (Bagratuni et al. 2010). High levels of spliced XBP1 mRNA were consistently detected in all 253 newly diagnosed MM patients, and high ratios of spliced versus unspliced XBP1 mRNA (>1.33) directly correlated with lower overall survival. Recent study showed that low XBP-1 levels predicted poor response to bortezomib, both in vitro and in MM patients. Moreover, selected bortezomib resistance MM cell lines down-regulate XBP-1 and immunoglobulin secretion (Ling et al. 2012). These findings suggest that XBP1 might play an important role in MM pathogenesis. Indeed, transgenic expression of XBP1s in mice also leads to plasma cell dyscrasia with evidence of increased monoclonal antibodies (‘M-spike’), lytic bone lesions, plasmacytosis and kidney damage (Carrasco et al. 2007). This study shows that XBP1 overexpression alone can drive transformation of plasma cells and promote multiple myeloma pathogenesis, underscoring the importance of dysregulated UPR in malignancy.
12.2.2 ATF6 and Multiple Myeloma
12.2.2.1 Overview
Among the three ER stress sensors, ATF6 does not dimerize to potentiate enzymatic activity. Instead, under ER stress conditions, ATF6 translocates to the Golgi apparatus and it is processed by site 1 protease (S1P) and site 2 protease (S2P) to release an active form of ATF6 (ATF6f). ATF6f translocates to the nucleus and activates target genes (Chen et al. 2002). In this capacity, ATF6 works in partnership with IRE1, as one of the target genes of ATF6 is XBP1, the key substrate of IRE1 (Yoshida et al. 2001). In addition to fueling the IRE1 arm of the ER stress response, ATF6 also functions as a transcription factor for ER chaperone proteins, thereby easing ER burden (Arai et al. 2006). These contributions to the ER stress response complement IRE1 activation and are generally adaptive, allowing such upregulation of protein production as is seen in plasma cell development. However, prolonged ATF6 activation can also result in transcription of CHOP, another transcription factor which enacts a largely apoptotic program of gene expression (Matsumoto et al. 1996). This effect of ATF6 activity occurs in conjunction with PERK activation, in contrast to the protective program that ATF6 and IRE1 jointly support.
One group has made an attempt in HeLa cells to describe the genetic modulation downstream of ATF6 activation and to distinguish it from the genetic signature of PERK activation (Okada et al. 2002). The group examined this question by comparing the cellular pool of mRNA in HeLa cells treated with the general ER-stress inducer tunicamycin with that of cells stably expressing the nuclear form of ATF6. From this experiment, the ATF6 contribution to the integrated ER stress response was extracted for HeLa cells. The primary targets identified were the expected ER chaperones grp78, gp96, and calreticulin (Okada et al. 2002). In addition, proteins which directly modify disulphide bonds to assure proper folding of nascent proteins were identified, such as ERp62 and ERp71 (Okada et al. 2002). Unfortunately, the authors concluded that this cell system was not conducive to the study of XBP1 transcription, which is critical for understanding myeloma development and progression. However, the research revealed that ATF6 and PERK both converge on CHOP transcription, confirming this as a locus of crosstalk between the two sensors (Okada et al. 2002).
CHOP (C/EBP homologous protein, alias GADD153) is a pro-apoptotic transcription factor routinely used as a read-out for activation of the ER stress response (Kawabata et al. 2012; Mimura et al. 2012; Schonthal 2013). The Mori group has proposed that CHOP transcription is most efficiently activated upon binding by both the nuclear form of ATF6 and ATF4, the transcription factor effector of PERK activation (Okada et al. 2002). The convergence of ER stress signals results in CHOP binding to its target genes, with inhibitory effects on some targets and transcriptional effects on others. CHOP activity results in the downregulation of the anti-apoptotic Bcl2 as well as the upregulation of the ER-resident oxidase ERO1-alpha (Marciniak et al. 2004). CHOP is also its own target, suggesting that its activation constitutes a commitment to programmed cell death (Marciniak et al. 2004).
12.2.2.2 ATF6 in MM
Surprisingly little has been reported about the role of ATF6 in MM, especially considering the important role it plays in the generation of the IRE1 substrate XBP1 (Lee et al. 2002). Indeed, the transcriptome of ATF6 should itself be a discrete target of research in the myeloma field.
Recent study showed that specific knockdown of ATF6 in MM cells resulted in significant cell death, and as is also the case for the other ER stress sensors (IRE1 and PERK) (Michallet et al. 2011). In addition, increased baseline signaling through the PERK sensor was enhanced upon knockdown of ATF6. Thus, the three sensors appear to all be required for baseline survival for MM cells, although crosstalk may allow for some limited compensation between the sensor systems.
Certainly, the crosstalk between ATF6 and the other two ER stress sensors suggests that ATF6 plays the role of a “swing vote.” When activated in conjunction with IRE1, growth and adaptation to protein production is reinforced. When linked to PERK, ATF6 activity can support an ER programmed cell death response. This duality indicates a potentially powerful target, identifying ATF6 as an understudied aspect of myeloma.
12.2.3 PERK and Multiple Myeloma
12.2.3.1 Overview
The Pancreatic eIF2-alpha kinase (PERK, alias EIF2αK3) is the third known sensor of ER stress and like the other two, it is embedded in the ER membrane. As the only such sensor left inactivated in the normal development of plasma cells, it has been of particular interest in the study of myeloma (Ma et al. 2010). We will therefore provide a summary of its canonical function and then review studies testing the role of PERK in baseline myeloma biology and in response to drug treatment.
Like the other two ER stress sensors, the activation of PERK requires its release by ER chaperone grp78. In addition, the other ER chaperone gp96 (alias grp94) has been shown to bind PERK at baseline and release it during ER stress conditions (Ma et al. 2002). Upon release, PERK is free to homodimerize and activate as a kinase. Active PERK has three interacting mechanisms, allowing gradations of cellular effects ranging from protective to destructive. These effects are mediated by eIF2-alpha, ATF4, and CHOP. First, the direct phosphorylation target of PERK is eIF2-alpha, a protein needed for ribosomal translation of mRNA (Wek and Cavener 2007). The phosphorylation of eIF2-alpha inhibits its activity, resulting in global repression of protein production. This strategy of translation repression reduces the load of nascent proteins being delivered to the ER for processing and is an effective short term answer to the problem of ER stress. However, the side effects of halting protein production are myriad, and the phosphorylation of eIF2-alpha does allow exceptions. For instance, mRNA with IRES sequences can still be translated under these conditions (Gerlitz et al. 2002). In addition, the transcription factor ATF4 is translated and subsequently translocated to the nucleus. The mechanism allowing such translation during eIF2α phosphorylation has been of significant interest and research has identified a double upstream open reading frame structure in the ATF4 mRNA which is preferentially translated when ribosomal processing is slowed (Kilberg et al. 2009; Lu et al. 2004). ATF4 then binds to genetic sequences with CCAAT motifs, many of which can be translated under the phosphorylated eIF2a condition which is downstream of PERK activation, likely due to upstream ORFs that function like the ones present in ATF4 mRNA (Kilberg et al. 2009; Lu et al. 2004). This activation of the ATF4 transcriptome is the second major arm of PERK response to ER stress.
ATF4 facilitates the transcription mRNAs coding for proteins with functions specific to ER stress conditions. For instance, redox-management genes are turned on, as well as additional chaperones for the ER (Harding et al. 2003; Liu et al. 2008; Ye and Koumenis 2009). Again, this strategy is adaptive for the cell and may allow the cell to cope with short term ER stress. However, the third arm of PERK signaling involves activation of CHOP, already described as a target of ATF6. The CHOP promoter includes binding sites for both ATF4 and ATF6, which appear to synergize (Okada et al. 2002). In addition, the CHOP mRNA includes an upstream inhibitory ORF that is preferentially translated during ER stress (Jousse et al. 2001; Lee et al. 2011). Expression of this protein is very tightly regulated and eventual convergence on CHOP activation signals a likely shift into macroautophagy and/or apoptosis (Emdad et al. 2011; Gomez-Santos et al. 2005; Kim et al. 2006). Thus, PERK has protective functions, especially when first activated, but it can also induce cell death pathways if it is too strongly activated or active for too long.
12.2.3.2 PERK as Protective Mechanism in MM
As previously referenced, Michallet et al. used RNAi to individually knock down IRE1, ATF6, and PERK expression (Michallet et al. 2011). They observed that loss of any one sensor tended to increase the activation read outs of the remaining sensors, confirming crosstalk between the systems. Specific knockdown of PERK yielded two important findings. First, this single change resulted in an autophagic cell death response, implicating PERK activation as a necessary part of the metabolic shift from plasma cell to myeloma cell. Second, the loss of PERK impeded the apoptotic response. Therefore, PERK activity was implicated in both viability of myeloma cells and in the apoptotic potential of the cells (Michallet et al. 2011). This complex finding may shed light on idea of PERK activity as a potential danger to the cell.
12.2.3.3 PERK as a Cell Death Effector in MM
Activation of PERK has been implicated in a wide variety of cancers as a mediator of response to chemotherapy (Fribley et al. 2011; Kraus et al. 2008; Lust et al. 2009; Qiao et al. 2012; Sailaja et al. 2013; Yan et al. 2010). Most convincingly, siRNA against PERK or dominant negative models can ameliorate chemotherapy-induced death in many types of cancer cells (Kahali et al. 2010; Lai and Wong 2008; Pan et al. 2012; Yacoub et al. 2008). It is therefore perhaps unsurprising that this effect has also been seen in myeloma cells, which already have baseline ER stress and may not be able to tolerate perturbations to the system. In particular, researchers have been interested in the role of PERK in myeloma cell response to the proteasome inhibitor bortezomib, the most effective myeloma therapy. Studies have demonstrated that bortezomib treatment upregulates PERK activity as measured by ATF4 and downstream CHOP expression (Obeng et al. 2006). Furthermore, they correlated ER stress to bortezomib response by measuring the retention of immunoglobulin protein accumulating in treated cells. Myeloma cells that retained more of their secretory protein load, one hallmark of ER stress, showed more activation of ER stress markers and more sensitivity to the drug (Obeng et al. 2006).
This pathway has been further probed in myeloma cells by induction of ER stress through inhibition of heat shock proteins, the family of ER chaperones that includes both grp78 and gp96. Most commonly, heat shock protein 90 is targeted experimentally with the drug 17-AAD. By comparison of 17-AAG and bortezomib effects on myeloma cells, the study showed that both drugs induce upregulation of grp78, gp96, and CHOP, all of which are downstream effects of PERK activation (Davenport et al. 2007). These effects were ultimately joined by an apoptotic response, suggesting that PERK activation culminated in a cell death program (Davenport et al. 2007).
The key component of apoptosis-induction by PERK was investigated to better understand the unfortunate phenomenon of bortezomib resistance in myeloma. Studies demonstrated that the phosphorylation of eIF2α is an indispensable aspect of PERK-mediated apoptosis (Schewe and Aguirre-Ghiso 2009). They studied a bortezomib-resistant subpopulation of myeloma cells and found that resistance could be reversed by inhibition of the eIF2α phosphatase or by competitive inhibition of the phosphatase via overexpression of a mutant phosphorylated eIF2α. In both conditions, cells with experimentally enhanced levels of endogenous phosphorylated eIF2α regained sensitivity to bortezomib (Schewe and Aguirre-Ghiso 2009).
The global repression of protein translation has far-reaching consequences, even if subsets of mRNAs are selectively processed. For instance, the balance of proteins in the cell quickly changes as proteins with short half-lives are degraded but not replaced. One system affected by such a change is the anti-apoptotic network, comprised of such anti-apoptotic proteins as survivin, Mcl-1, and FLIP, all of which are eliminated from the protein pool if not continuously generated (White-Gilbertson et al. 2009). This time-dependent shift in cellular fitness may be another axis on which PERK activation is titrated, so that short term activation is beneficial while long-term activation is ultimately detrimental to the cell.
12.3 Targeting UPR Pathways and HSPs Against MM: The Opportunities
UPR plays critical roles in plasma cell differentiation as well as in the pathogenesis of multiple myeloma (MM). XBP1s and downstream ER chaperones are consistently up regulated in myeloma cells in patients (Bagratuni et al. 2010), inspiring increasing efforts to develop UPR-targeted anti-MM therapy (Mimura et al. 2012; Papandreou et al. 2011; Ri et al. 2012).
12.3.1 Blockade of IRE1/XBP1s in MM
In addition to the critical roles of IRE1/XBP1 in plasma cell differentiation, a picture has emerged for the roles of UPR in myeloma. Indeed, XBP1s and downstream ER chaperones are consistently up regulated in myeloma patients. Patients with a low XBP1 spliced/unspliced ratio (≤1.33) have a longer overall survival compared with those with a higher ratio (p = 0.03, median, 56 months vs 40 months; HR = 1.75; 95 % CI = 1.07–2.85) (Bagratuni et al. 2010). Moreover, transgenic expression of XBP1s in mice also leads to plasma cell dyscrasia with evidence of increased monoclonal antibodies (“M-spike”), lytic bone lesions, plasmacytosis and kidney damage (Carrasco et al. 2007). Given this information, IRE1/XBP1 could be a potential therapeutic target for multiple myeloma.
To investigate whether blocking the IRE1/XBP1 pathway is a therapeutic for MM, researchers performed chemical library screening and identified a small-molecule compound, STF-083010, which specifically blocks the endonuclease activity of IRE1 without affecting its kinase activity (Papandreou et al. 2011). Furthermore, treatment of a variety of myeloma cell lines with STF-083010 in vitro demonstrated its potent activity against myeloma. Importantly, STF-083010 is also selectively more cytotoxic to freshly isolated CD138+ plasma cells from myeloma patients than CD19+ B cells, CD3+ T cells and CD56+ NK cells. Finally, treatment of human myeloma xenografts in NSG mice was performed. STF-083010 was given by intraperitoneal injection on day 1 and day 8 and this compound significantly inhibited the growth of these tumors in vivo (Papandreou et al. 2011). In addition, another small-molecule inhibitor, MKC-3946 also blocks the IER1α endoribonuclease domain. MKC-3946 inhibits multiple human myeloma cell lines without toxicity to normal mononuclear cells. MKC-3946 also blocks ER stress induced by both bortezomib and heat shock protein 90 inhibitor 17-AAG. Moreover, MKC-3946 can significantly enhance the cytotoxicity of MM cells induced by bortezomib or 17-AAG (Mimura et al. 2012). A similar result was found by using an XBP1 inhibitor, toyocamycin, which was identified from the culture broth of an Actinomycete strain. Toyocamycin has been shown to suppress the XBP1 mRNA splicing in HeLa cells which is induced by thapsigargin, tunicamycin, and 2-deoxyglucose. It does not, however, affect ATF6 and PERK activation. Although toyocamycin does not inhibit IRE1α phosphorylation, it prevents IRE1α-induced XBP1 mRNA cleavage and inhibits constitutive activation of XBP1 expression in myeloma cell lines as well as in samples from myeloma patients in vitro. Toyocamycin also induces apoptosis of myeloma cells, including bortezomib-resistant myeloma cells, and it inhibits myeloma cell growth in a human myeloma xenograft model (Ri et al. 2012). Taken together, these results demonstrate that blockade of IRE1/XBP1 pathway by small-molecule compounds is a potentially useful therapeutic modality for human myeloma (Table 12.1).
Table 12.1
Summary of targeting of UPR pathways in MM
Agent | Target | Effect on UPR and MM | Phase | References |
|---|---|---|---|---|
Toyocamycin | IRE1α-XBP1 | Induce XBP1s, not affect ATF6 and PERK | Preclinical studies | Ri et al. (2012) |
Induce apoptosis of MM cells, including bortezomib-resistant cells in vitro, and also inhibit MM cell growth in vivo | ||||
STF-083010 | IRE1α | Block the endonuclease activity without affecting kinase activity | Preclinical studies | Papandreou et al. (2011) |
Inhibit MM cell growth in vitro and in vivo | ||||
MKC-3946 | IRE1α | Block the endonuclease activity | Preclinical studies | Mimura et al. (2012) |
Inhibit MM cell growth in vitro and in vivo. Blocks ER stress induced by bortezomib and HSP90 inhibitor 17-AAG |
12.3.2 Targeting HSPs Is a Promising Therapeutic Platform for MM
Heat shock proteins are a group of highly conserved proteins, which are involved in many cellular processes such as protein folding, intracellular trafficking, modulating signaling pathways and regulating immune responses. Heat shock proteins play critical roles in the regulation of protein homeostasis and cell survival. Inhibition of HSPs results in a disruption of protein processing and induces ER stress. Therefore, HSPs are attractive targets for treatment of MM (Table 12.2).
Table 12.2
Summary of targeting of HSPs in MM