- 15.1 Introduction
- 15.2 Mechanisms of endocrine autoimmunity
- 15.3 Thyroid disease
- 15.3.1 Thyrotoxicosis
- 15.3.2 Hashimoto’s thyroiditis
- 15.3.3 Idiopathic thyroid atrophy (myxoedema)
- 15.3.1 Thyrotoxicosis
- 15.4 Diabetes mellitus
- 15.4.1 Classification of diabetes mellitus
- 15.4.2 Immunopathogenesis of Type 1 diabetes mellitus
- 15.4.3 Treatment of diabetes mellitus
- 15.4.4 Complications of diabetes mellitus
- 15.4.5 Are immunological tests useful?
- 15.4.1 Classification of diabetes mellitus
- 15.5 Adrenal disease
- 15.5.1 Autoimmune adrenal disease
- 15.6 Parathyroid disease
- 15.7 Gonadal disease
- 15.7.1 Oophoritis
- 15.8 Infertility
- 15.8.1 Immunology of infertility
- 15.9 Pituitary disease
- 15.10 Autoimmune polyendocrine disease
 Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
15.1 Introduction
Endocrine cells may be localized in a defined glandular structure such as the adrenal gland, or distributed throughout a non-endocrine organ such as the stomach. Functional disorders of endocrine glands result from overactivity, with excessive production of the hormone, or from atrophy, with failure to produce the relevant hormone. There are many causes of glandular dysfunction but autoimmunity to endocrine tissues is one of the commonest.
Most autoimmune endocrine disorders are clinically silent until they present with features of insufficiency of the affected organ. At this stage the gland is often irreversibly damaged with little prospect of recovery even if the autoimmune process were arrested. Current treatment of many of these diseases therefore centres on replacement of hormones. The long period of silent inflammation and glandular destruction, which can last for many years, offers a window during which progress of these diseases could potentially be reversed, if this were detected. However, detection and treatment of preclinical endocrine autoimmunity is currently confined to experimental studies involving small numbers of first-degree relatives of subjects with polyendocrine syndromes or autoimmune diabetes, to define those who are at increased risk of developing the condition.
15.2 Mechanisms of endocrine autoimmunity
Autoimmune reactions may be directed against endocrine cells, their receptors, hormones or the receptors on the cells targeted by the hormone (Fig. 15.1).
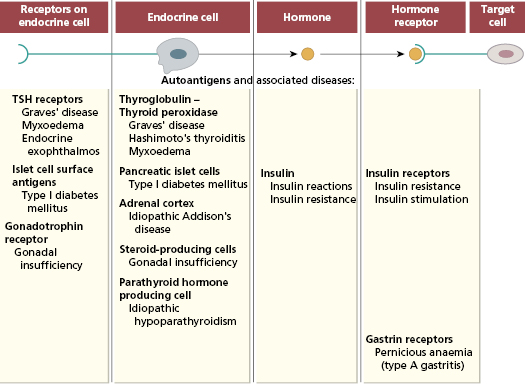
Since the first report of autoimmune thyroid disease in 1957, autoimmune diseases of every endocrine organ and virtually all endocrine cells have been described.
Autoantibodies to endocrine cells are organ specific and detected only by tests using antigen from the specific endocrine gland involved (Chapter 19). This contrasts with non-organ-specific diseases (such as systemic lupus erythematosus), where non-organ-specific antigens (such as nuclear antigens) are present in all organs and tissues of the body. A feature of organ-specific autoimmunity is that several autoantibodies to endocrine glands may be found in a single patient; this patient may have clinical evidence of one or many endocrine disorders or may be asymptomatic; in other words, the presence of autoantibodies does not necessarily indicate autoimmune disease.
There are several mechanisms of autoimmune damage (Fig. 15.1) and more than one mechanism may occur in a given disease. Evidence suggests that both T cells and antibodies often work in parallel to produce autoimmune endocrine disease. As a broad generalization, T cells (both CD4 and CD8) are responsible for glandular destruction and antibodies act via mechanisms discussed in section 15.10 to disturb the physiological function of the gland. Primary autoantibodies are pathogenic and act on surface receptors of the target cells; those autoantibodies that are secondary to glandular destruction acting as markers of disease. Alternatively, neutralisation of enzymatic activity by autoantibodies may be pathogenic too.
Antibodies can influence the function or growth of an endocrine gland. Primary stimulating and blocking antibodies against receptors are well recognized, as are antibodies that selectively influence cell growth. Patients may have a mixture of receptor antibodies, some of which stimulate and some of which block the receptor. Shifts from one type to the other explain why some patients fluctuate from over-activity to under-activity of the gland.
15.3 Thyroid disease
Several thyroid antigens are recognized, including the TSH receptor, thyroglobulin, thyroid peroxidase (thyroid microsomal antigen), sodium/iodide symporter (the transporter responsible for iodine uptake by thyroid cells), as well as other surface and cytoplasmic thyroid antigens. The most widely available and clinically useful antibody tests are those for thyroid peroxidase (secondary or marker antibodies) and the TSH receptor (primary pathogenic antibodies) (Table 15.1).
Table 15.1 Prevalence and relative strength of antibodies to thyroid peroxidase commonly detected in various thyroid diseases
| Clinical presentation | Antibodies to thyroid peroxidase |
|---|---|
| Thyrotoxicosis | |
| Graves’ disease | Positive (low titre) |
| Hot nodules | Negative |
| Goitre | |
| Hashimoto’s thyroiditis | Positive (high titre) |
| Simple goitre | Negative |
| De Quervain’s thyroiditis | Transient positive |
| Carcinoma | Negative |
| Thyroxine deficiency | |
| Primary myxoedema | Positive |
| Normal population | Positive (5–10%) |
15.3.1 Thyrotoxicosis
Thyrotoxicosis is a common condition with a prevalence of about 20 per 1000 of the population. It can occur at any age, but the incidence peaks in the third and fourth decades. It is about five to 10 times more common in women than men. Thyrotoxicosis is most commonly due to Graves’ disease (autoimmune hyperthyroidism) or to local hyperactive nodules in the thyroid gland. The diagnosis is made by biochemical thyroid function tests (see Case 15.1). Serum autoantibodies to thyroid peroxidase confirms Graves’ disease but these antibodies occur in other autoimmune thyroid conditions too (see Table 15.1). Those patients who have high titres of these autoantibodies are the ones most likely to proceed to thyroid failure known as myxoedema.
 Case 15.1 Graves’ disease
Case 15.1 Graves’ diseaseA 29-year-old woman presented with a 3-month history of increased sweating and palpitations with weight loss of 7 kg. On examination, she was a nervous, agitated woman with an obvious, diffuse, non-tender, smooth enlargement of her thyroid, over which a bruit could be heard. She had a fine tremor of her fingers and a resting pulse rate of 150/min. She had no evidence of exophthalmos. A maternal aunt had suffered from ‘thyroid disease’.
On investigation, she had a raised serum T3 of 4.8 nmol/l (NR 0.8–2.4) and a T4 of 48 nmol/l (NR 9–23). Measurement of her thyroid-stimulating hormone showed that this was low normal, 0.4 mU/l (NR 0.4–5 mU/l). The biochemical findings pointed to primary thyroid disease rather than pituitary overactivity. Circulating antibodies to thyroid peroxidase (titre 1/3000; 200 IU/ml) were detected by agglutination. A diagnosis of autoimmune thyrotoxicosis (Graves’ disease) was made. She was treated with an antithyroid drug, carbimazole, to control her thyrotoxicosis, and surgery was not required.
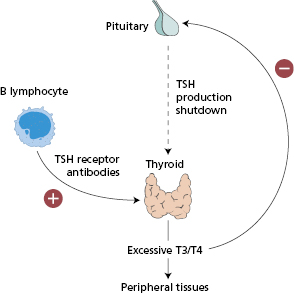
The pathogenesis involves IgG autoantibodies to the TSH receptor on the surface of human thyroid cells (Fig. 15.2). Almost all patients with Graves’ disease have TSH receptor antibodies that stimulate the thyroid cell (thyroid-stimulating antibodies) (Figs 15.2 and 15.3), resulting in thyrotoxicosis or overactive thyroid; however, it is not necessary to measure these autoantibodies routinely for diagnosis.
Fig. 15.2 Surface of a thyroid cell showing actions of the primary antibodies in autoimmune thyroid diseases. LATS, Long-acting thyroid stimulator.
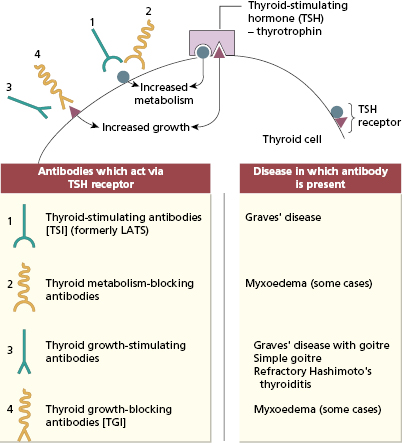
The autoimmune thyroid is characteristically infiltrated by T lymphocytes: both CD8+ and CD4+ cells are present. These T cells express a more limited number of T-cell receptor genes (see Chapter 1) than do peripheral blood T cells from the same patient; the implication is that intrathyroid T cells are less diverse because they are enriched for T cells specific for thyroid-derived peptides.
Two of every 1000 pregnant women are thyrotoxic and occasionally such pregnancies result in neonatal Graves’ disease. This is due to transplacental transfer of thyroid-stimulating IgG autoantibodies from mother to fetus. The neonatal disease can be severe; affected babies have goitre, exophthalmos, feeding problems, pyrexia and tachycardia and may develop heart failure unless treated promptly. Spontaneous recovery gradually occurs over 2–3 months, as the maternal IgG is metabolized at a rate consistent with its half-life (i.e. 3–4 weeks) (see Chapter 18).
The degree of thyrotoxicosis in Graves’ disease is not related to the size of the goitre; indeed, 10% of patients do not have an enlarged thyroid. Thyroid growth-stimulating immunoglobulin (TGI) has been demonstrated in the sera of patients with Graves’ disease with goitre, as well as in some patients with toxic multinodular goitres or non-toxic goitres. In contrast to the thyroid-stimulating immunoglobulins (TSI) which cause hyperthyroidism, these antibodies correlate with goitre size but not with the overproduction of T3 and T4.
Half of the patients with Graves’ disease develop exophthalmos; this may precede, coincide with or follow the hyperthyroid phase. It may even occur occasionally in euthyroid patients or in association with Hashimoto’s thyroiditis or primary myxoedema. Smoking is a strong risk factor. Exophthalmos results from myositis affecting the eye muscles and a proliferation of retro-orbital tissue. The myositis is accompanied by infiltration of lymphocytes – particularly CD4+ T cells. Retro-orbital fibroblasts secrete hydrophilic glycosaminoglycans (GAGs) in response to T-cell cytokine signalling, altering the osmotic pressure and resulting in fluid accumulation. The sera from affected patients contain antibodies that bind to eye muscle extract; some of these antibodies cross-react with other orbital antigens as well as thyroid antigens. The TSH receptor seems to be aberrantly expressed in the inflamed orbital tissue, suggesting that an immune response to this antigen may also contribute to the eye disease. In patients with severe exophthalmos and optic nerve compression (‘malignant exophthalmos’), high-dose steroids are of value, sometimes coupled with other immunosuppressive drugs. If there is deterioration, surgical decompression is indicated.
A few (3–5%) patients with Graves’ disease develop pretibial myxoedema; they tend to have exophthalmos as well. Pretibial myxoedema refers to well-demarcated, subcutaneous thickening of the anterolateral aspects of the legs; these areas do not pit on pressure, and are shiny and reddish brown in appearance. The development of pretibial myxoedema is not related to the duration or extent of the hyperthyroidism. Its pathogenesis is not well understood, although, as in thyroid eye disease, aberrant expression of the TSH receptor has been described in the affected tissue and secretion of GAGs by cytokine-stimulated fibroblasts is believed to play a role.
Genetic factors are important in the aetiology of Graves’ disease. A positive family history of hyperthyroidism is found in around 50% of patients and there is 20–40% concordance in monozygotic twins but less than 5% in dizygotic twins. As with several other organ specific autoimmune diseases, HLA-DR3 and polymorphisms of the CTLA-4 gene (see Chapter 1) are both strongly associated with Graves’ disease in Caucasians but genetic associations in other ethnic groups are less well defined. Environmental triggers of Graves’ disease remain obscure. Some limited evidence exists for infection with several viruses in thyroid tissue in patients with the disease. There is also an association between the onset of Graves’ disease and psychological stress. Treatment of multiple sclerosis with the lymphocyte-depleting monoclonal antibody Campath-1H can induce Graves’ disease in around 10% of those treated, possibly by depleting an inhibitory T-cell population (see Chapter 5).
Graves’ disease can be treated successfully by antithyroid drugs or surgery. Immunosuppressive therapy to reduce levels of the causative antibodies has not therefore proved necessary (Box 15.1).
- Thyroid infiltration by T lymphocytes (both CD4+ and CD8+ cells) and plasma cells
- The presence of circulating autoantibodies to thyroid antigens, especially the TSH receptor. Antibodies to the TSH receptor cause stimulation of cultured thyroid cells
- An increased risk of thyroid disease in first-degree relatives of patients with Graves’ disease
- Associations with other autoimmune diseases, including myasthenia gravis, pernicious anaemia and rheumatoid arthritis
- Transient Graves’ disease in the neonates of pregnant women with Graves’ disease
15.3.2 Hashimoto’s thyroiditis
Hashimoto’s disease is much more common in women than in men and is probably the commonest cause of goitre in the UK. At presentation, 75% of patients are euthyroid, 20% are hypothyroid, and the remaining 5% are hyperthyroid and have a disease that closely resembles Graves’ disease (known as ‘Hashitoxicosis’). About 50% of patients eventually become hypothyroid due to destruction of the thyroid gland. Hashimoto’s thyroiditis is familial and associated with other organ-specific autoimmune diseases. The close relationship and familial association with Graves’ disease, combined with the fact that Hashimoto’s thyroiditis may sometimes evolve in to Graves’ disease (and vice versa), implies a linked pathogenesis.
 Case 15.2 Hashimoto’s thyroiditis
Case 15.2 Hashimoto’s thyroiditisA 39-year-old woman presented with a large, painless swelling in her neck. The enlargement had been a gradual process over 2 years. She had no other symptoms and felt generally well. On examination, her thyroid was diffusively enlarged and had a rubbery consistency. There were no signs of thyrotoxicosis or of thyroid failure on clinical examination.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


