Hormone
Dose at which risk of dysfunction is increased
Endocrine consequence
Recommendations for screening and evaluation as clinically indicated
GH
≥18 Gy
• Growth hormone deficiency
• Growth velocity
• Poor linear growth (may be masked by concomitant CPP)
• Tanner staging
• Shorter adult height
• Bone age
• IGF-1
• GH provocative testing
LH/FSH
≥18 Gy
• CPP (girls <age 5 most susceptible); rapid linear growth (may be masked by concomitant GH deficiency)
• Growth velocity
• Premature menarche
• Tanner staging
• Shorter adult height
• Pelvic ultrasound (girls)
• Bone age
• LH/FSH and testosterone/estradiol
• Leuprolide stimulation test
LH/FSH
≥30
• Hypogonadotropic hypogonadism
• Growth velocity
• Delayed/incomplete puberty
• Tanner staging
• Shorter adult height
• Bone age
• LH/FSH, PRL, and testosterone/estradiol
TSH
≥30
• Central hypothyroidism
• Free T4 and TSH
• Shorter adult height
ACTH
≥30
• Central adrenal insufficiency
• 8 AM cortisol
• Adrenal crisis
• Cosyntropin stimulation test
PRL
≥50
• Galactorrhea
• Prolactin
• Hypogonadotropic hypogonadism
Data suggest that age has a differential effect on susceptibility of the HPA to damage from irradiation (Darzy 2009). The risk of developing GH deficiency, in particular, has been associated with younger age at treatment by most but not all authors (Diller et al. 2009; Chemaitilly and Sklar 2010; Darzy 2009; Chemaitilly et al. 2015a; Mulder et al. 2009; Brauner et al. 1986; Schmiegelow et al. 2000a).
Another important predisposing factor for injury to the HPA is the dose of irradiation. As would be predicted, the higher the radiation dose, the more likely the child is to develop endocrine sequelae (Chemaitilly and Sklar 2010; Tonorezos et al. 2015; Darzy 2009; Chemaitilly et al. 2015a; Mulder et al. 2009; Rutter and Rose 2007; Laughton et al. 2008; Merchant et al. 2011). Cranial radiotherapy doses ≥18 Gy impart a significant risk for GH deficiency, which can be an isolated event; doses ≥30 Gy increase the risk of LH/FSH, ACTH, and TSH deficiencies; and radiation doses exceeding 50 Gy are more likely to be associated with multiple anterior pituitary hormone deficiencies and hyperprolactinemia. Conversely, there is no exact threshold dose below which there is no risk of HPA injury, as GH deficiency can even be observed in children who receive low-dose (8–11 Gy) irradiation (Mulder et al. 2009). Although the use of proton therapy is better suited to spare the HPA when it is distal to the desired radiation target, well-designed studies comparing protons versus photons are lacking, although a recent cohort-simulation model would suggest that proton therapy may be more cost-effective for scenarios in which radiation dose to the hypothalamus can be spared (Mailhot Vega et al. 2015).
Third, the gender of the patient is another variable that may play a role in the risk of radiation-induced neuroendocrine dysfunction. Males appear to be at increased risk for any endocrine complication in general (Brignardello et al. 2013), and men may be at higher risk for LH/FSH deficiencies (Chemaitilly et al. 2015a). As also seen in the non-tumor population, girls are more likely to get central precocious puberty (CPP) (Darzy 2009; Chemaitilly et al. 2015b). Hyperprolactinemia also occurs more commonly in women (Darzy 2009; Constine et al. 1993).
Finally, the time elapsed since radiation treatment is an important determinant of the likelihood of having an endocrine disorder. Studies have documented that the cumulative incidence of a chronic endocrine condition continues to rise even decades after diagnosis (Brignardello et al. 2013; Diller et al. 2009). The same is true for HPA dysfunction, which may take years to evolve, especially with lower radiation doses (Chemaitilly and Sklar 2010; Tonorezos et al. 2015; Darzy 2009; Chemaitilly et al. 2015a; Merchant et al. 2011; Gleeson and Shalet 2004). Notably, the onset of pituitary dysfunction after irradiation can also occur within just a few years (Laughton et al. 2008), especially with higher radiation doses (Merchant et al. 2011).
31.3 Pathophysiology
The pathophysiology of radiation-induced injury to the HPA remains poorly understood, but it is believed that neuroendocrine dysfunction most likely arises from direct neuronal injury, although there may also be a contribution from vascular damage and reduced blood flow (Chieng et al. 1991). The hypothalamus is believed to be the site of the HPA most impacted by irradiation (Gleeson and Shalet 2004). This is based on the observation of a preserved GH response to exogenous GH-releasing hormone in GH deficient patients exposed to cranial radiation and because of the known occurrence of hyperprolactinemia following high-dose radiotherapy to the CNS (Constine et al. 1993; Schriock et al. 1984; Lam et al. 1986; Schmiegelow et al. 2000b). There are, nevertheless, data supporting a certain degree of vulnerability of the pituitary gland to radiation, especially with higher radiation doses (Darzy 2009; Gleeson and Shalet 2004). It is therefore safe to assume that HPA dysfunction after irradiation most likely results from a combination of events at both the hypothalamic and pituitary levels.
In general, there is a hierarchy of anterior pituitary hormone loss after radiation therapy. GH is the most frequently impacted followed by LH/FSH, TSH, ACTH, and usually after much higher radiation doses, PRL (Brignardello et al. 2013; Gurney et al. 2003a; Darzy 2009; Chemaitilly et al. 2015a; Langsenlehner et al. 2007; Littley et al. 1989; Gan et al. 2015) (Table 31.1). As a rule, the posterior pituitary (the distal axons of hypothalamic neurons that synthesize arginine vasopressin and oxytocin) is not injured by radiation therapy, and hence, central diabetes insipidus is not a frequent or expected late effect of CNS irradiation but is more likely to result from direct tumor involvement or surgery (Brignardello et al. 2013; Miyoshi et al. 2008; Darzy 2009; Gleeson and Shalet 2004; Lam et al. 1986; Langsenlehner et al. 2007).
31.4 Clinical Presentation and Consequences
As discussed above, radiation-induced HPA dysfunction may occur years after the completion of therapy, and it is an evolutionary process that rarely encompasses all HPA hormones simultaneously from the outset. Because the onset of radiation-induced hypopituitarism is usually insidious, it may be difficult to diagnose. The symptoms may be mild and overlooked, especially in survivors who are coping with multiple treatment-induced morbidities. In addition, the presentation of multiple endocrinopathies, such as the coexistence of GH deficiency and CPP (Figs. 31.1 and 31.2), may make the clinical diagnosis challenging. The consequences of neuroendocrine dysfunction can have a profound long-term impact on the cancer survivor, including shorter adult height (that may be disproportional [Fig. 31.2]), obesity, low bone mass, a higher burden of chronic disease, increased frailty, and lower quality of life (Diller et al. 2009; Chemaitilly et al. 2015a).
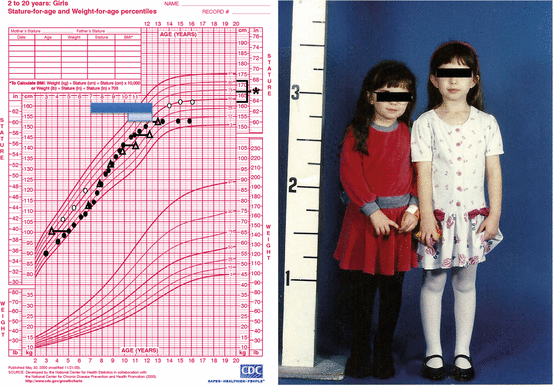
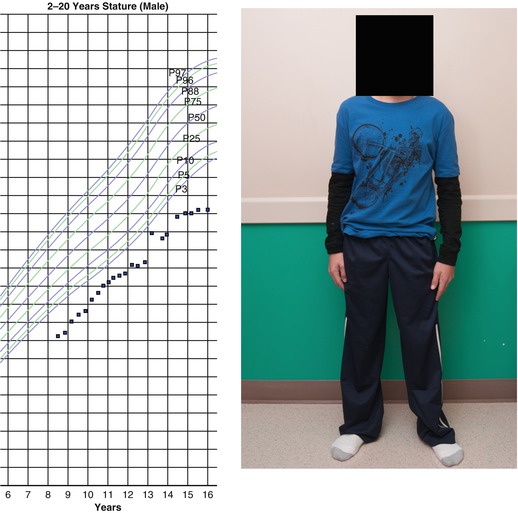
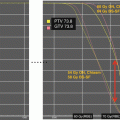
Fig. 31.1
Growth chart of a girl (solid circles) with a history of locally advanced retinoblastoma status post right eye enucleation, chemotherapy, and radiotherapy (39.6 Gy in 35 fractions) to the orbit, completed at the age of 17 months. Bone ages are represented by open triangles. She was diagnosed with growth hormone (GH) deficiency and then central precocious puberty and treated with recombinant human GH (dark blue box) and depot leuprolide (light blue box), respectively. The patient had menarche at age 8 and achieved a final height of 60 in., well below mid-parental height (asterisk). At age 23 years, she was diagnosed with hypogonadotropic hypogonadism. The patient’s identical twin sister (open circles) was already significantly taller by the age of 5 years. She had menarche at age 12 and achieved a final adult height of 63.75 in. (Figure reproduced with permission from (Stephen et al. 2011))
Fig. 31.2
A 15-year-old boy with a history of medulloblastoma diagnosed at age 14 months who ultimately received craniospinal irradiation (30 Gy with an additional 25.2 Gy boost to the posterior fossa) at the age of 5 due to recurrent disease. He was diagnosed with and initiated therapy for GH deficiency at the age of 8 and, at age 10 years, he started depot leuprolide due to early pubertal development. Despite these interventions, he had an extremely short final adult height and disproportionate stature (trunk disproportionately short compared with limbs)
31.4.1 GH Deficiency
GH deficiency (GHD) is the most common, and often the only, anterior pituitary hormone deficit following radiotherapy that includes the HPA in the radiation field. In a recent study of 748 long-term adult survivors of childhood cancer treated with cranial radiotherapy (including survivors of acute lymphoblastic leukemia and CNS tumors), the prevalence estimate of GHD was 46.5% (95%CI, 42.9–50.2%) (Chemaitilly et al. 2015a). The risk of GHD is even higher in cohorts of brain tumor survivors (Laughton et al. 2008). Based on the results of one study in 192 pediatric patients with localized brain tumors, a cumulative radiation dose of 16.1 Gy to the hypothalamus was determined to be the mean dose required to achieve a 50% risk of GHD at 5 years (Merchant et al. 2011).
Despite being common, however, GHD is not the only contributor to poor linear growth during childhood, especially during active cancer therapy when confounding factors such as poor nutrition and the use of glucocorticoids may also be present. Years after radiation therapy, concomitant endocrinopathies such as hypothyroidism and pubertal disorders may also affect height and growth velocity. Furthermore, final adult height in children with GHD may be negatively impacted by having received concomitant spine radiotherapy, which can directly damage the vertebral epiphyses and lead to disproportionate growth (upper/lower segment ratio < 0.9; arm span more than 5 cm > height; sitting height standard deviation < standing height standard deviation) (Fig. 31.2) (Diller et al. 2009; Shalet et al. 1987; Clayton and Shalet 1991; Edgar et al. 2009; Meacham 2003).
The clinical presentation and diagnosis of GHD is reviewed in more detail elsewhere and is primarily based on auxological criteria (Growth Hormone Research Society 2000; Stanley 2012). In general, GHD classically manifests as a subnormal growth velocity (normal is >5 cm/year in prepubertal children) associated with a crossing of height percentiles. The bone age X-ray (plain radiograph of the left arm and wrist that assesses skeletal maturity via a comparison of the epiphyses with age-matched standards) is typically delayed for the child’s chronologic age, and the insulin-like growth factor 1 (IGF-1) and IGF-binding protein 3 (IGF-BP3) are characteristically low for age and pubertal status. GH provocative testing is frequently utilized to confirm a diagnosis of GHD, and in fact is often required by third-party payors despite issues with reliability and reproducibility.
There are some notable exceptions regarding the presentation of GHD in the child who has received irradiation. First is that plasma IGF-1 and IGF-BP3 levels are not always reliable screening tests for GHD in children exposed to CNS radiation as they may be normal in individuals with a diagnosis proven by provocative testing (Weinzimer et al. 1999). Second, GHD may occur concomitantly with premature sexual maturation in a large number of pediatric cancer survivors (Figs. 31.1 and 31.2) (Sklar and Constine 1995), and therefore the bone age may not be delayed or may even be advanced; obesity and previous chemotherapy exposure (e.g., treatment with retinoic acid) may also influence the bone age in this fashion (Hobbie et al. 2011; Russell et al. 2001). Third, growth velocity may be normal in the child with profound GHD and concomitant CPP or obesity (Edgar et al. 2009). Finally, GHD may be partial and may be manifested only by a diminished pubertal growth spurt (Meacham 2003).
The treatment of GHD in childhood generally involves the use of daily subcutaneous injections of recombinant human GH, which in turn can improve final height outcome (Diller et al. 2009; Gleeson et al. 2003; Ciaccio et al. 2010), although children treated with radiotherapy may never fully achieve their genetic height potential (Beckers et al. 2010). Given the proliferative and pro-mitogenic properties of GH and IGF-1, the safety of GH therapy in individuals with a history of cancer has been an area of concern. Recent studies have not identified a clear risk from GH therapy, specifically as relates to the development of second CNS neoplasms (Patterson et al. 2014), and current understanding is that it is safe to prescribe GH to the pediatric cancer survivor as early as 1 year after cancer therapy is complete (Raman et al. 2015).
31.4.2 Central Precocious Puberty and LH/FSH Deficiencies
Puberty is characterized by the development of sex-specific secondary sexual characteristics, accelerated growth, and ultimately fusion of the growth plates and cessation of linear growth. In girls, breast development is the first sign of puberty whereas in boys, it is testicular enlargement with the possible and notable exception of those concurrently exposed to gonadotoxic therapies. CPP, commonly defined as pubertal onset before age 8 in girls and 9 in boys, arises from the premature activation of the hypothalamic-pituitary-gonadal axis and, if untreated, may cause a final adult height significant shorter than is genetically predetermined. LH and FSH deficiencies become apparent when the child has delayed entrance into puberty (>age 13 in girls or >age 14 in boys), delayed menarche (>age 16 in girls), or develops hypogonadotropic hypogonadism (amenorrhea in girls, low testosterone levels in boys with inappropriately low or normal LH/FSH levels) after puberty has begun. The diagnosis and treatment of pubertal disorders is more comprehensively reviewed elsewhere (Chen and Eugster 2015; Palmert and Dunkel 2012).
Cranial radiation is a significant risk factor for dysfunctional gonadotropin secretion (Diller et al. 2009; Laughton et al. 2008; Gleeson and Shalet 2004; Gan et al. 2015; Edgar et al. 2009; Meacham 2003; Stephen et al. 2011; Oberfield et al. 1996; Armstrong et al. 2009a). CPP may occur following treatment with a wide range of cranial radiotherapy doses, especially >20–25 Gy (Gleeson and Shalet 2004; Muller 2002; Ogilvy-Stuart et al. 1994), with younger age at radiotherapy conferring the highest risk of CPP (Oberfield et al. 1996; Ogilvy-Stuart et al. 1994; Lannering et al. 1997). Patients who develop CPP following radiation doses exceeding 30 Gy have a significant risk of ultimately developing LH/FSH deficiencies (Constine et al. 1993; Toogood 2004; Brauner et al. 1984) (Fig. 31.1). The timing of menarche can also be impacted by irradiation, especially when treatment is received at ages younger than age 5, and both early and late menarche can occur (Armstrong et al. 2009b; Chow et al. 2008).
In pediatric cancer survivors, the clinical presentation of CPP can pose distinct challenges. As mentioned above, CPP can mask co-occurring GHD by inducing seemingly normal growth rates at the expense of rapid bone age advancement, leading to reduced adult height (Figs. 31.1 and 31.2). Importantly, increasing testicular volume, which is the hallmark of pubertal entrance in boys, may not be reliable in the identification of pubertal onset in male childhood cancer survivors who received cytotoxic chemotherapy or direct testicular radiation; thus a high index of suspicion and measurement of LH/FSH and testosterone levels supplement the physical exam (Armstrong et al. 2009a; Quigley et al. 1989). Plasma estradiol and gonadotropin levels and/or uterine length measurements using pelvic ultrasound may likewise be helpful in determining the pubertal status of a subset of girls.
31.4.3 TSH Deficiency
Deficiency of TSH secretion results in insufficient stimulation of thyroid follicular cells and resultant low circulating thyroxine levels, which can cause typical signs and symptoms of hypothyroidism in children, including decreased growth velocity, delayed skeletal maturation, constipation, skin dryness, and constitutional symptoms such fatigue and cold intolerance. The main risk factor associated with TSH deficiency is HPA exposure to radiation doses ≥30 Gy (Chemaitilly et al. 2015a; Laughton et al. 2008; Rose et al. 1999). As there is no widely available provocative testing, the diagnosis of central hypothyroidism is made by documenting a serum-free thyroxine level below the lab-specific normal range in the presence of a low, inappropriately normal, or minimally elevated serum TSH level. The evolution of TSH deficiency can be prolonged and the diagnosis difficult to make; often, the downward trend in thyroxine levels can help to alert the clinician to the diagnosis (Darzy 2009).
31.4.4 ACTH Deficiency
The hypothalamic-pituitary-adrenal axis, although more resilient, can also be damaged by the effects of irradiation. ACTH deficiency causes low serum cortisol levels through gradual atrophy of the adrenal cortex, and central adrenal insufficiency can present with symptoms of anorexia/nausea, weakness, fatigue, and unintentional weight loss. Aldosterone production is not under the direct control of ACTH and so hyperkalemia and salt wasting are not components of ACTH deficiency although symptomatic hypotension can occur during episodes of acute physiological stress when the compensatory secretion of cortisol fails to occur. The main risk factor for central adrenal insufficiency is HPA exposure to radiation doses exceeding 30–40 Gy (Chemaitilly et al. 2015a; Laughton et al. 2008; Rose et al. 2005; Patterson et al. 2009). The diagnosis of ACTH deficiency is usually made after cosyntropin stimulation (Chrousos et al. 2009), as random morning plasma ACTH and cortisol levels may be unreliable (Patterson et al. 2009). ACTH deficiency can also be partial, and some patients may require only stress steroid coverage instead of commencing daily glucocorticoid replacement (Darzy 2009). Although one of the least common endocrine sequelae of CNS irradiation, central adrenal insufficiency is nonetheless an important entity to recognize given the potential life-threatening consequences of untreated cortisol deficiency, especially during acute illness.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


