Introduction
The human body maintains homeostasis via multiple regulatory systems that exert tight control over every organ system, with continuous regulation via negative feedback mechanisms. These systems often have overlapping and complementary effects leading to coordinated changes across multiple body systems in response to a stimulus or stressor. Heart rate, blood pressure, and systemic venous and arterial tone are controlled via the sympathetic and parasympathetic nervous systems, interfacing closely with renin, aldosterone, angiotensin, and arginine vasopressin (AVP) to regulate intravascular volume and osmolality. The hypothalamus–pituitary–adrenal axis is responsible for ensuring appropriate adrenergic and glucocorticoid responses to stress, with downstream effects on metabolisms, glycolysis, gluconeogenesis, and immune function. These systems work in concert to maintain homeostasis in response to stressors including exercise, illness, and injury. Under ideal conditions, with resolution of the external stressor, these systems return to baseline function and homeostasis is restored.
Critical illness, by definition, is any life-threatening condition that would result in death without support of one or more organ systems. During critical illness, homeostasis is severely disrupted, and one or more regulatory systems are unable to compensate for the inciting event. When regulatory mechanisms are inadequate or, conversely, inadequately regulated, they can cause or exacerbate critical illness. For example, sepsis is defined as a dysregulated host response to infection resulting in end organ dysfunction. Additionally, critical illness, especially when prolonged, can result in uncoupling of normal stimulus-response homeostatic mechanisms, resulting in prolonged derangement in physiologic parameters and the inability to return to a baseline state of health, described as chronic critical illness. Although a consensus definition for chronic critical illness does not yet exist, it includes patients with a prolonged (more than 7 days ) illness who suffer ongoing organ dysfunction, cognitive decline, malnutrition and an ongoing catabolic state, recurrent or persistent infections, and profound persistent weakness.
In this chapter we provide an overview of the normal (expected) response to critical illness and trauma, review what is known about endocrine systems and regulation during chronic critical illness, and discuss specific systems which are impacted during these events. Understanding normal and dysregulated host responses to critical illness and trauma is a vital part of providing critical care, and investigation into the mechanisms underlying chronic critical illness represents a rich target for future interventions in patients with this condition.
Normal Response to Stress
SYMPATHETIC NERVOUS SYSTEM
Stimulation of the sympathetic nervous system due to pain, fever, injury, and illness results in stimulation of the adrenal medulla, which releases epinephrine, and post-ganglionic cells of the sympathetic nervous system, which release norepinephrine. Levels of these hormones can rise 10- to 100-fold in the setting of an acute stress response. Norepinephrine primarily stimulates α-receptors, whereas epinephrine stimulates both β- and α-receptors, with multiple effects throughout the body. Alpha-receptor activation results in vasoconstriction and increased vascular tone and relaxation of smooth muscle of the gastrointestinal tract. Beta-receptor activation results in bronchodilation, as well as increased heart rate (chronotropy) and contractility (inotropy), which work in concert to increase cardiac output. Collectively, these actions work to increase oxygen delivery and vascular tone.
Some patients exhibit a reduced response to increases in epinephrine and norepinephrine in acute illness or trauma. This is especially common in elderly adults and is multifactorial in nature. Resting sympathetic tone increases with increasing age, concomitant with decrease sensitivity of α- and β-receptors, resulting in decreased response to elevated sympathetic nervous system output. Arterioles in older adults are less responsive to α-adrenergic stimulation, and older adults have a reduced maximal heart rate. In addition, older patients are more likely to be prescribed medications such as beta-blockers and calcium channel blockers that prevent increased vasoconstriction and chronotropy. In these patients, the pulse may be inappropriately normal, and early signs of hypovolemia and/or sepsis may be easily missed. Additionally, these patients have reduced physiologic reserve, as they are unable to mount an adrenergic response commensurate with an inciting stressor and can quickly develop hypotension and malperfusion as a result of vasoplegia and hypovolemia. Clinicians should have a high index of suspicion when evaluating these patients and be alert for signs of hypovolemia, hemorrhage, and sepsis.
RENIN–ANGIOTENSIN–ALDOSTERONE SYSTEM
In concert with tight management of serum osmolality and total body volume, regulated by AVP (see later), the renin-angiotensin-aldosterone system is responsible for managing sodium balance and extracellular sodium levels via several mechanisms. Renin is secreted by juxtaglomerular cells in the afferent arteriole of the kidney in response to decreased perfusion pressure, activation of sympathetic nerve fibers in the afferent arterioles, and decreased NaCl delivery to the macula densa. Renin then activates angiotensinogen, produced in the liver, to yield angiotensin I, which moves through the circulation where it is converted to angiotensin II by angiotensin-converting enzyme by pulmonary and renal endothelial cells. Angiotensin II stimulates aldosterone release by the adrenal cortex which enhances NaCl reabsorption throughout the proximal and distal tubule and collecting duct, increases arteriolar vascular tone, which acts to increase blood pressure, and stimulates AVP secretion and decreases urine water losses to increase circulating volume.
ARGININE VASOPRESSIN
The pituitary is closely coupled with the renal system to maintain water balance, serum osmolarity, and intravascular volume. AVP is responsible for regulating the amount of water excreted into urine, therefore maintaining appropriate body water volume and serum osmolality. The strongest mediator of AVP secretion is serum osmolality. In the setting of appropriate serum osmolality, serum AVP levels are low, and the renal collecting duct excretes high volumes of water, resulting in urine with low osmolality. However, as the serum osmolality climbs, relatively small changes in serum osmolality substantially increase AVP secretion. Cells of the anterior hypothalamus called osmoreceptors sense the rising osmolality and signal production of AVP in the supraoptic and paraventricular nuclei of the hypothalamus, which is transported in granules to the posterior pituitary before being released. Serum AVP travels to the renal system where it induces the retention of free water, reducing urine production and returning serum osmolality to normal levels, and returning AVP production to baseline levels.
AVP is also regulated to a lesser extent by decreased blood pressure or blood volume. This is driven by baroreceptors in both low-pressure left atrium and pulmonary artery, which respond to changes in vascular volume, and high-pressure baroreceptors in the aortic arch and carotid sinus, which respond to changes in arterial pressure. Signals from these baroreceptors are relayed to the supraoptic and paraventricular nuclei of the hypothalamus, resulting in AVP production and release. However, these baroreceptors are less sensitive than the osmoreceptors regulating serum osmolality, and a 5% to 10% decrease in blood volume or blood pressure is required before AVP will be released by this mechanism. Additionally, changes in blood pressure and blood volume alter the hypothalamus’s response to changes in serum osmolality. With hypovolemia, small changes in osmolality will cause AVP release at osmolalities previously within the normal range. This will result in the conservation of intravascular volume at the expense of decreased serum osmolality. Conversely, with blood volume overload, the serum osmolality set point shifts in the opposite direction, and a higher osmolality is required before AVP will be secreted, resulting in increased water diuresis.
Critically ill patients may develop two conditions involving dysregulation of AVP and serum osmolality. Patients may over-secrete AVP in syndrome of inappropriate anti-diuretic hormone, or SIADH, resulting in inappropriate water retention, low urine output, and hyponatremia. This condition can be caused by malignancy, trauma including intracranial hemorrhage, pulmonary conditions including pneumonia, and medications including selective serotonin reuptake inhibitors (SSRIs), thiazide diuretics, and antiepileptic medications including carbamazepime and oxcarbazepine. Diagnostic criteria include serum Na less than 135 mmol/L, serum osmolality less than 275 mOsm/kg, and spontaneous urine osmolality greater than 100 mOsm/kg. Treatment typically consists of fluid restriction and addressing the underlying etiology causing SIADH.
Conversely, central diabetes insipidus (DI) results from the lack of production and release of AVP. When this occurs, there is minimal water reabsorption in the renal collecting ducts and patients can produce up to 20 L of dilute urine per day, resulting in substantial hypovolemia and hypernatremia. Central DI is commonly caused by traumatic brain injury, central nervous system infections, and tumors. Diagnosis is obtained by performing a water deprivation test, with measurement of serum and urine osmolality before and after water deprivation. Treatment involves reversing inciting causes, when possible, and administration of desmopressin, a vasopressin analogue, to stimulate renal water reabsorption.
In addition to its role in regulating serum osmolality and intravascular volume, AVP also plays a key role in maintaining vascular tone under normal conditions and when patients are in shock. Several conditions, including sepsis and hemorrhagic shock, disrupt normal vasomotor tone, resulting in vasoplegia, hypotension, and hypoperfusion. This can occur despite activation of catecholamine release and activation of the renin-angiotensin system and is mediated at least in part due to increased production of nitric oxide. , Vasopressin levels would be expected to increase with hypovolemia and hypotension; however, as shock worsens, plasma vasopressin levels decrease, , perhaps due to depletion of vasopressin stores in the neurohypophysis. Restoration of normal vasopressin levels may improve outcomes in patients with shock: in a recent randomized clinical trial, Sims et al. found that low-dose supplementation of AVP in patients with traumatic hemorrhage had decreased transfusion requirements. A separate randomized controlled trial (RCT) by Gorden et al. found that patients with septic shock required less renal replacement therapy when randomized to vasopressin (0.06 units/minute) versus epinephrine, but no difference in survival.
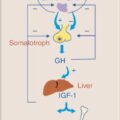
GROWTH HORMONE
Growth hormone (GH) is released by the anterior pituitary in a pulsatile fashion in response to hypothalamic release of growth hormone-releasing hormone (GHRH). GH has complex effects on multiple peripheral tissues, but in general promotes protein anabolism by increasing amino acid uptake and increased protein synthesis, and utilization of fat stores by stimulating triglyceride breakdown. Regulation of GH release is complex and can be stimulated by hormones other than GHRH, including ghrelin, and inhibited by somatostatin and insulin-like growth factor 1 (IGF-1). In critical illness, GH secretion increases, with higher peaks and more frequent pulses of hormone secretion, stimulating release of IGF-1 and inducing lipolysis and hepatic gluconeogenesis.
In chronic critical illness, GH release pulsatility is suppressed, correlating with low levels of IGF-1, and appears to contribute to muscle and bone loss seen in chronic critical illness. ,
HYPOTHALAMIC–PITUITARY–ADRENAL AXIS
Stressful stimuli activate the hypothalamic–pituitary–adrenal (HPA) axis. The hypothalamus releases corticotropin-releasing hormone (CRH), which stimulates the pituitary to release adrenocorticotropic hormone (ACTH). ACTH in turn stimulates the adrenal cortex to release cortisol, a steroid hormone with numerous effects throughout the body. Both cortisol and cortisone have a much higher affinity for mineralocorticoid receptors than glucocorticoid receptors, and at baseline levels of hormone, largely bind to mineralocorticoid receptors. However, with stress, glucocorticoid levels increase and begin occupying glucocorticoid receptors as well. Glucocorticoids, after binding to their receptors in the cellular cytosol, pass into the cell nucleus to regulate transcription of genes related to metabolism, inflammation, and the immune response.
Regulation of the HPA Axis
Under normal circumstances, the HPA axis operates via negative feedback. Elevations in ACTH and cortisol result in decreased secretion of CRH and ACTH, respectively. With acute critical illness, cortisol concentrations are elevated substantially above baseline and remain elevated for several days. With moderate and short episodes of stress and subsequent glucocorticoid secretion, negative feedback inhibition of the HPA axis is rapid (minutes to hours). Several studies have found that ACTH levels in critically ill patients were substantially lower than baseline, consistent with negative feedback. Despite this, cortisol levels remain elevated, due in part to decreased cortisol degradation. , , In experimental studies, critically ill patients had a cortisol half-life five times longer than non-critically ill patients. , Interpretation of serum cortisol levels is complicated by the fact that most cortisol is bound to cortisol-binding globulin and therefore not biologically active; furthermore, stress states appear to impact the availability of glucocorticoid receptors, further modulating the effects of the elevated cortisol levels seen in illness and injury.
With ongoing illness and glucocorticoid production, inhibition of the HPA axis decreases and the normal circadian variation of cortisol is flattened, promoting skeletal muscle catabolism and muscle wasting with fat redistribution, central adiposity, and development of metabolic syndrome and insulin resistance or type 2 diabetes. With prolonged use, corticosteroids also decrease osteoblast number and function resulting in decreased bone formation, osteopenia and osteoporosis, and frequently results in hypertension by causing increased vascular tone in response to baseline levels of endogenous vasoactive agents.
Glucocorticoids in Acute Critical Illness
Glucocorticoids play several critical roles in the normal stress response. They increase vascular reactivity to other vasoactive agents, including norepinephrine and angiotensin II. Glucocorticoids also decrease overall inflammation via several mechanisms. They reduce cell apoptosis at sites of injury and inflammation while simultaneously inducing apoptosis of inflammatory immune cells, including eosinophils, thymocytes, mast cells, and antigen-presenting cells such as dendritic cells. Inhibition of antigen presentation results in a lymphocyte shift from T helper 1 (Th1) to T helper 2 (Th2) cells, shifting cytokine production away from interferon (IFN)-γ, interleukin (IL)-2, and tumor necrosis factor (TNF)-β and encouraging production of IL-4, IL-5, IL-6, IL-10, and IL-13. Overall, this dampens the adaptive immune response and works to prevent an over exuberant host immune response to infection, which could result in excessive hypotension, inflammation, and end-organ dysfunction, which contribute to sepsis. Concomitant with this dampening effect, hematopoietic stem cells are induced by IL-1, IL-6, and other cytokines to produce populations of immature myeloid cells. This myelopoiesis seems to balance the dampening of the adaptive immune response by bolstering the innate immune system.
Glucocorticoids also regulate metabolism via several mechanisms. Glucocorticoids induce inhibition of glucose uptake in peripheral tissues including skin, fibroblasts, white blood cells, and adipocytes, resulting in peripheral hyperglycemia and relative insulin resistance. Glucocorticoids also stimulate lipolysis and muscle cell release of amino acids and activate glucose-6-phosphatase. Together, these effects result in increased hepatic gluconeogenesis by making substrates for gluconeogenesis available and increasing the enzymes necessary for this process. Finally, glucocorticoids induce hepatic liver glycogen synthesis via increased synthesis and activation of hepatic glycogen synthase. This mechanism can be interpreted as preparation for rapid production of glucose on demand from glycogen stores. Overall, these metabolic changes result in decreased lipogenesis and muscle anabolism and increased gluconeogenesis and glycogen storage, resulting in the increased glucose availability for tissues during times of stress.
Glucocorticoids in Chronic Critical Illness
In chronic critical illness, ACTH levels remain suppressed, but cortisol levels remain persistently elevated due to reduced degradation and loss of binding proteins, including albumin, resulting in ACTH-cortisol dissociation. With minimal ACTH stimulation, adrenal tissue atrophies, resulting in depletion of cholesterol esthers and the enzymes necessary for epinephrine and norepinephrine production.
Ongoing glucocorticoid-driven myelopoiesis results in ongoing immunosuppression, which is perpetuated by myeloid-derived suppressor cells that secrete antiinflammatory cytokines IL-10 and transforming growth factor (TGF)-β, antagonize clonal lymphocyte expansion, and decrease T-lymphocyte responsiveness, the persistence of which is associated with poor outcomes in patients with sepsis, , , , including increased risk of nosocomial infection, prolonged intensive care unit (ICU) stay, and mortality. Ongoing marrow production of myeloid cell populations comes at the expense of lymphopoiesis and erythropoiesis, resulting in immunosuppression and anemia or chronic disease. This prolonged state of simultaneous immunosuppression and inflammation has been termed the “Persistent Inflammation-Immunosuppression Catabolism Syndrome” and is typified by a patient admitted with sepsis or severe injury who has ongoing organ failure, persistent malnutrition and protein catabolism despite appropriate supplementation of macronutrients and calories, poor wound healing, and ongoing functional decline.
Inappropriate or Inadequate Response to Stress
ADRENAL INSUFFICIENCY
Humans and animals without functional adrenal glands are unable to cope with increased metabolic demands from acute illness or injury. Animals and human patients with adrenal insufficiency demonstrate reduced efficacy in the setting of vasoactive medications, which resolves with administration of exogenous glucocorticoids.
Absolute adrenal insufficiency can result from bilateral adrenalectomy, adrenal hemorrhage or trauma, autoimmune conditions, and metastases to the adrenal glands with tissue destruction and subsequent loss of function. These patients frequently have symptoms of adrenal insufficiency in day-to-day life requiring steroid supplementation. More commonly in the ICU, critically ill patients demonstrate a relative adrenal insufficiency in which they are unable to respond appropriately to an acute stressor with an appropriate increase in cortisol production. Relative adrenal insufficiency should be suspected in patients receiving prolonged glucocorticoid therapy due to chronic suppression of ACTH and subsequent adrenal atrophy and in patients with peripheral vascular collapse despite appropriate volume resuscitation. Acute adrenal insufficiency is discussed further in Chapter 14.
THYROIDAL DISEASE
The thyroid is controlled by the hypothalamic–pituitary–thyroid (HPT) axis and feedback loop. The hypothalamus releases thyrotropin-releasing hormone (TRH), which stimulates the secretion of thyroid-stimulating hormone (TSH) by the anterior pituitary. TSH then causes release of thyroid hormones from the thyroid gland. Thyroxine (T4) is converted to the active hormone triiodothyronine (T3). Studies have shown that serum concentrations of these hormones are adaptive and respond to certain environmental factors, and therefore are impacted by critical illness.
Patients hospitalized in the ICU often have low serum concentrations of T4 and T3. In this patient population, despite low T3 and T4, TSH is typically maintained within normal range or is slightly decreased. This is a deviation from the typical negative feedback regulation in the HPT axis, which in primary hypothyroidism would result in an increase in serum TSH. These laboratory value changes found in patients with critical illness are referred to as nonthyroidal illness syndrome (NTIS).
Evidence suggests that sickness, including both acute and chronic illnesses, result in a decrease in serum T3 concentration. As disease progresses, serum T3 further decreases, and is associated with prognosis. NTIS is thought to be present in the majority of critically ill patients. Debate exists as to whether this hypothyroidism is protective or harmful to their recovery. For example, many patients in the ICU seem to display the signs and symptoms classic to patients with hypothyroidism, including hypothermia, muscle weakness, impaired consciousness, and depressed myocardial function. On the other hand, the patient may benefit from depressed metabolism in order to avoid uncontrolled catabolism.
The pathogenesis of NTIS is not entirely understood. The HPT axis is downregulated by severe illness at both the hypothalamic and pituitary level, causing subsequent changes to the negative feedback regulation in the HPT axis. Additionally, liver metabolism of thyroid hormone, as well as the enzymatic processes associated with thyroid hormone metabolism, are altered. Likewise, the proinflammatory cytokines involved in the acute phase response of critical illness directly affect thyroid hormone metabolism. Unlike T4, which is produced only in the thyroid, 80% of T3 is produced by the peripheral 5′-doidination of T4 to T3 in muscle, liver, and kidney by 5′-monodeiodinases (D1 and D2). During critical illness, T3 production decreases. Several mechanisms are thought to inhibit D1 and D2, such as high serum cortisol concentration, high circulating free fatty acid, treatment with drugs that inhibit 5′-monodeiodinase activity, like amiodarone and propranolol, and elevated levels of cytokines.
An additional factor affecting thyroid function in the ICU is poor oral and enteral intake, resulting in malnutrition. Decreased caloric intake during critical illness is thought to contribute to the development of NTIS. Serum thyroid hormones are decreased during a period of fasting and during decreased caloric intake in patients with critical illness. Likewise, many of the drugs commonly used in critical illness result in decreased thyroid function. For example, patients receiving high-dose glucocorticoids (greater than 20 mg/day prednisone), dopamine, or dobutamine often have suppressed TSH. However, in patients with hyperthyroidism, TSH is classically less than 0.01, whereas patients receiving the medications listed above typically have values in the range of 0.08 to 0.4. Therefore, a sensitive TSH assay can be useful. Other frequently used medications, including furosemide, nonsteroidal antiinflammatory drugs (NSAIDs), heparin, anticonvulsants, and metformin, can alter thyroid function tests by mechanisms that are not completely understood.
Patients who take levothyroxine for known hypothyroidism should have it continued during their hospitalization. Likewise, patients with long-standing hypothyroidism and new-onset critical illness are at increased risk of the development of myxedema coma. Given the prevalence of NTIS in patients with critical illness and the decrease of serum thyroid hormones (especially T3) in this patient population, the diagnosis of untreated hypothyroidism can be challenging. In patients suspected to have primary hypothyroidism, the most useful test is measurement of plasma TSH. This is because a normal plasma TSH will rule out primary hypothyroidism. Patients with NTIS who also have primary hypothyroidism will have a high serum TSH. Of note, patients with acute illness and primary hypothyroidism may have a decrease in the typical rise in TSH. Therefore, in this patient population, a high serum TSH and low serum T4 is indicative of hypothyroidism; however, these laboratory abnormalities have been seen in patients recovering from NTIS. Primary hypothyroidism is also supported by a high serum T3 to T4 ratio and typically this ratio is reversed in NTIS. The treatment of hypothyroidism and myxedema coma is to replace thyroid hormone, treat underlying illnesses, and provide supportive care. ,
Diagnosis of hyperthyroidism in patients with NTIS is important and can be difficult, as patients in the ICU are not immune to thyrotoxicosis or thyroid storm. Furthermore, surgery and infection are known precipitating factors of thyroid storm, and therefore patients in the ICU are at increased risk. The combination of decreased TSH, high T4, and normal T3 may suggest a combination of thyrotoxicosis and NTIS but are not specific. Symptoms of thyroid storm, including arrhythmias, fever, and altered mental status, should raise the suspicion of hyperthyroidism in the critically ill. Patients with presumed thyrotoxicosis and thyroid storm in the ICU should be treated aggressively with supportive measures, beta-blockers, intravenous glucocorticoids, and antithyroid drugs. Thyrotoxicosis is discussed further in Chapters 1 and 2.
The outcomes of critically ill patients with abnormal thyroid function tests have been studied but are largely inconclusive. For example, a low serum T3 is associated with increased hospital stay and need for mechanical ventilations for patients with acute heart failure. Additionally, low serum T3 is predictive of a 30-day mortality in patients with community-acquired pneumonia. Low serum T4 values are also associated with increased mortality in critically ill patients. However, additional evidence suggests that low serum T3 levels may be beneficial in the critically ill.
As mentioned previously, the treatment of NTIS is the ICU is controversial. Only a few, small RCTs have studied the effects of treatment with thyroid hormones on patients with NTIS, and there is little consistency regarding treatment choice, as both T3 and levothyroxine have been studied. Additionally, it is noteworthy that normalizing serum thyroid hormone concentrations do not necessarily result in normal tissue concentrations of thyroid hormones in the critically ill. One subset of patients who may benefit from thyroid hormone treatment is patients with heart failure. Although there has been largely inadequate study on this subject, the results are encouraging. An RCT of patients with dilated cardiomyopathy showed positive effects of levothyroxine treatment on cardiac function. In general, there is no conclusion regarding the effectiveness of thyroid hormone treatment of patients in the ICU with NTIS.
In chronic critical illness, hypothalamic TRH is decreased, TSH secretion pulsaltility is lost, and patients demonstrate low plasma T4 and T3. Although the exact mechanism driving TRH suppression is unclear, it appears to be due to an altered set point for feedback inhibition within the hypothalamus. Multiple animal and human studies demonstrate peripheral tissue adaptation by increasing thyroid hormone transporters and increasing peripheral activation of thyroid hormone. These low peripheral T3 levels correlate with muscle and bone anabolism, contributing to the ongoing metabolic wasting seen in the chronically critically ill. ,
GLYCEMIC DYSREGULATION
Glycemic dysregulation, often referred to as “stress hyperglycemia” in the critically ill and trauma population, is common and associated with many factors. The pathophysiology is multifactorial and only partially understood. During critical illness, complex interactions between counterregulatory hormones and cytokines cause excessive production of glucose, which is also coupled with insulin resistance. Stress increases glycogenolysis and gluconeogenesis. Glycogenolysis is triggered by increased catecholamines, whereas gluconeogenesis is triggered by an increase in stress response glucagon. Additionally, insulin resistance exacerbates hyperglycemia and is described as the inability of muscle and adipocyte tissue to take up glucose, which is caused by an alteration of insulin signaling and the downregulation of type 4 glucose transporters (GLUT-4) during critical illness. Likewise, post injury or postoperatively, the influence of cytokines can induce insulin resistance. Additionally, increased glucose reabsorption and decreased renal glucose clearance has been described. All of these factors together lead to glycemic dysregulation and hyperglycemia in the trauma and critically ill patient population.
In the past, hyperglycemia was not routinely treated in the ICU, as it was considered an essential adaptive response to critical illness. However, uncontrolled hyperglycemia has been studied extensively and is associated with poor outcomes. As a result, greater attention is paid to the correction of hyperglycemia in the ICU. In a large RCT of 1826 patients, patients in the ICU with hyperglycemia had a higher mortality that those who were normoglycemic. Several subgroups of critically ill patients have been studied as well. For example, hyperglycemic patients following trauma have increased mortality, length of stay, and incidence of infection. , Similarly, patients with isolated traumatic brain injury and hyperglycemia have worse neurologic outcomes and increased intracranial pressure.
Although it is clear that preventing uncontrolled hyperglycemia in the critically ill and trauma population is desirable, the optimal blood glucose range remains controversial. A single-center RCT of 1548 ICU patients known as the Leuven surgical trial compared conventional blood glucose management (targeted blood glucose of 180 to 200 mg/dL) with intention-to-treat (IIT) (targeted blood glucose of 80 to 110 mg/dL) and found that overall mortality was improved in the IIT group. However, this study was never able to be replicated and received criticism for an abnormally high mortality rate among the control group. In a later large, multicenter trial, the Normoglycemia in Intensive Care Evaluation Survival Using Glucose Algorithm Regulation (NICE-SUGAR) trial, surgical patients treated with IIT (target blood glucose level of 81 to 108 mg/dL) had significantly increased risk of severe hypoglycemia and increased 90-day mortality when compared with the conventional glucose control group (target blood glucose of less than 180 mg/dL). In addition, the Volume Substitution and Insulin Therapy in Severe Sepsis (VISEP) trial was a multicenter trial that found that ITT (target blood glucose level of 80 to 110 mg/dL) significantly increased the rates of hypoglycemia as well as serious adverse events when compared with conventional glucose control (target blood glucose level of 180 to 200 mg/dL).
In support of this evidence, as well as additional studies in critically ill patients, a targeted blood glucose of 140 to 180 mg/dL in most critically ill adult patients is recommended, rather than a more stringent target of 80 to 110 mg/dL or a more liberal target of 180 to 200 mg/dL. In order to achieve this target, intravenous fluids containing glucose should be minimized. Additionally, attempts should be made to avoid prolonged hypoglycemia. Short-acting insulin and insulin infusions should commonly be employed in the ICU to avoid this complication. ,
Summary
Patients with chronic critical illness experience derangements in multiple endocrine signaling pathways. Clinicians caring for these patients must have a firm understanding of the normal endocrine responses to critical illness and injury, as well as the ability to identify when these derangements are inadequate, maladaptive, and require additional therapy. The remainder of this book will expand on these conditions, providing a framework for diagnosis and management.
Substantial work remains to be done to improve our understanding of both normal endocrine feedback loops as well as the role of endocrine dysregulation in the development of chronic critical illness. Future work must identify the mechanisms that allow some patients to recover from critical illness and injury while others remain susceptible to ongoing infection, inflammation, and ongoing decline. Ultimately, intervening in these system derangements offers the possibility of improving outcomes for patients who survive their acute illness, decreasing the incidence of chronic critical illness and persistent inflammation-immunosuppression catabolism syndrome.
References
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


