Pathology
Penetrance
Endocrine
Parathyroid
> 95 %
Enteropancreatic
Gastrinoma
30–40 %
Insulinoma
10 %
Functional
2 %
Nonfunctional
20 %
Pituitary
Prolactinoma
20 %
Other functional
10 %
Nonfunctional
5 %
Foregut neuroendocrine
Gastric
10 %
Thymic
6 %
Bronchial
3 %
Adrenal
Nonfunctioning
20–30 %
Pheochromocytoma
< 1 %
Non-Endocrine
Facial angiofibromas
85 %
Collagenomas
70 %
Lipomas
10–30 %
Leiomyomas
10 %
Meningiomas
5 %
Ependymomas
1 %
Patients with MEN-1 have lower life expectancy with a 50 % mortality rate by the age of 50 years [35–37]. Death in MEN-1 is most commonly due to the malignant progression of pancreatic disease [35, 37]. While parathyroid and pituitary lesions can cause significant morbidity, they account for a smaller proportion of the overall mortality rate.
The main principles of managing patients with MEN-1 are to treat the hormone excess and preventing malignant progression of tumors. These are discussed below within each affected organ system.
Parathyroid Tumors
Clinical Presentation
Primary HPT is the most common clinical manifestation in individuals with MEN-1. Studies have shown that by 40 years of age, approximately 95 % of patients with MEN-1 mutations have parathyroid disease [27, 30]. HPT is also the most common initial presentation of MEN-1, accounting for 60–90 % of presenting complaints. The rate of symptomatic hypercalcemia is markedly decreased to 30 % in those patients within a known MEN-1 kindred who are known to be at risk and are undergoing regular biochemical surveillance. MEN-1 HPT accounts for only 2–4 % of all HPT cases in the community and 10 % of those with multiglandular disease [38]. MEN-1-related HPT differs from sporadic cases, and these differences have implications for the surgical management of this disease. First, the typical age of presentation is approximately 20–25 years of age, which is in marked contrast to sporadic HPT, which most commonly occurs during the fifth decade of life [27, 30]. Second, the sex distribution for MEN-1-related HPT is equal with males just as likely to have HPT as females [30, 32]. In contrast, sporadic HPT is three times more common in females. Thirdly, the pattern of parathyroid enlargement differs between MEN-1 and sporadic HPT. In sporadic HPT, approximately 90 % of cases are due to a single adenoma, while, in comparison, HPT in MEN-1 presents with multiglandular disease [39, 40]. The development of parathyroid hyperplasia in MEN-1 is asynchronous in nature with all four glands ultimately involved but at varying times. This gives rise to a pattern of asymmetrical disease with some glands showing significant growth, while others appear initially to be macroscopically normal [40, 41]. MEN-1 parathyroid disease is associated with supernumerary glands and ectopic glands in up to 15 % [42–44]. This pattern of multiglandular involvement and frequent occurrence of extra glands supports the traditional surgical approach of four-gland parathyroid exploration with cervical thymectomy. This approach has been challenged more recently with some groups advocating focused parathyroidectomy in selected cases [45]. Histopathologically, the enlarged glands show multiple monoclonal tumors, which have developed from polyclonal hyperplasia [46]. MEN-1 patients appear to demonstrate markedly increased levels of parathyroid mitogenic factor found within the serum of MEN1 patients. This mitogenic factor is similar to fibroblast growth factor and influences growth of the endothelial component of parathyroid glands [47]. Despite this mitogenic influence, it is unusual for HPT cases in MEN-1 to progress to parathyroid cancer [48–52].
Aside from the differences in age and sex distribution at presentation, the symptoms of MEN-1-associated HPT do not differ significantly from those seen in the sporadic form of the disease. In the past, up to 50 % of MEN-1 cases have presented with symptomatic renal calculi; however, with the advent of genetic testing and aggressive screening protocols, more patients are being managed with subclinical disease. The typical symptoms of HPT including bone abnormalities, musculoskeletal complaints, fatigue, and change in mental state are also seen. MEN-1 patients with HPT may complain of fatigue, polydipsia, polyuria, joint pain, bone pain, constipation, and sometimes depression.
Diagnosis
Inappropriately elevated intact serum parathyroid hormone levels in conjunction with hypercalcemia (raised ionized or total-albumin corrected serum calcium) are the principal biochemical characteristic in the diagnosis of HPT. Biochemical confirmation of HPT should always include measurement of 24-h urinary calcium to exclude benign familial hypocalciuric hypercalcemia. Determination of 1,25-dihydroxyvitamin D3 levels and serum blood urea and nitrogen and glomerular filtration rate (GFR) are important to rule out secondary causes of HPT. Screening for other elements of the MEN-1syndrome in patients considered a risk such as gastrin, insulin, Chromogranin A, pancreatic polypeptide (PP), IGF-1, VIP, glucagon, and prolactin can be obtained. Screening also includes cross-sectional imaging of the pancreas, adrenal glands, thymus, and pituitary. With the advent of minimally invasive surgery and focused parathyroid exploration for sporadic HPT, localization studies with ultrasound, sestamibi scintigraphy, or 4-D CT (four-dimensional computerized tomography) are now well established in the preoperative workup of sporadic cases. However, the role of localization studies for parathyroid tumors in MEN-1 is more controversial. This is because HPT in MEN-1 is multiglandular and traditional surgical approaches have required bilateral exploration. Parathyroid imaging for preoperative localization has not traditionally been advocated as a thorough four-gland exploration is the most widely adopted surgical approach [26, 53, 54]. However, the frequency of supernumerary glands and ectopic parathyroid locations challenges this view, and our practice more recently has been to routinely perform ultrasound and sestamibi scintigraphy prior to parathyroid surgery in MEN-1. This approach may also provide the surgeon and patient with the option of undertaking focused or minimally invasive parathyroid surgery if the imaging points to enlarged parathyroid glands confined to just one side of the neck [45]. Careful preoperative imaging is very important in planning surgery for patients with recurrent or persistent HPT, and in this setting concordant functional and anatomical imaging should be sought.
Management
The indications for parathyroid surgery generally follow the guidelines suggested for the management of asymptomatic HPT [55]. MEN-1 patients with Zollinger–Ellison syndrome should be considered for early parathyroid surgery as hypercalcemia is a potent stimulus for gastrin secretion, and normalizing parathyroid hormone levels improves control of hypergastrinemia [56, 57]. With the advent of easily available hormone assays, DNA analysis, improved imaging modalities, and surveillance of known MEN-1 kindreds, patients are increasingly presenting with mild HPT and are often asymptomatic. The question in these cases is when to initiate surgical intervention.
The ideal surgical approach to parathyroid disease in MEN-1 should strike a balance between minimizing the incidence of persistent or recurrent disease and avoiding permanent hypoparathyroidism. Surgery should be initiated in a timely fashion prior to the development of significant renal or skeletal complications. Prophylactic parathyroidectomy is generally not supported, and it is important that the surgeon understands that the outcome of permanent hypoparathyroidism may be worse than the disease itself considering there is rarely any malignant potential to parathyroid tumors in MEN-1 HPT. The timing of surgery and the specific surgical approaches are important and debatable. Some groups have suggested that delaying surgery until hypercalcemia worsens or symptoms develop allows the pathological glands to enlarge making success at the first operation greater and reduces the need for subsequent reoperations. However, proponents for early surgical intervention argue that normalization of hypercalcemia may not only decrease gastrin production but also help to avoid the skeletal and renal complications of HPT [57–62].
The main operative approaches at initial surgery are total parathyroidectomy with autotransplantation of a portion of parathyroid tissue (total parathyroidectomy; tPTX) or subtotal parathyroidectomy (sPTX; Fig. 1). In both procedures, cervical thymectomy is recommended to allow inspection of the thymus for supernumerary glands as well as potentially reducing the chance of thymic carcinoid tumors developing. In a small number of cases where preoperative imaging suggests asymmetric parathyroid disease, and parathyroid enlargement is confined to one side of the neck, we have undertaken unilateral parathyroid exploration with removal of the ipsilateral thymus via a small cervical incision [45]. This approach certainly avoids the problem of hypoparathyroidism; however, its long-term durability requires longer follow-up.
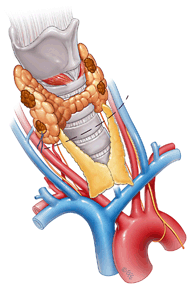
Fig. 1
Subtotal parathyroidectomy and bilateral cervical thymectomy. Incision is made over the dotted line. Then all four parathyroid glands are visualized with excision of all but approximately 50 mg of right inferior gland which is marked with suture. Bilateral cervical thymectomies are performed at the same time
The operative approach requires a delicate balance between avoiding recurrence and preventing hypoparathyroidism. The two main surgical approaches for MEN-1 HPT are tPTX or sPTX (Fig. 1). Procedures which involve a lesser approach to parathyroidectomy (< sPTX) than tPTX or sPTX are associated with higher (> 50 %) persistent or recurrent rates and lower disease-free intervals (7 years for < sPTX vs. 16.5 years for tPTX or sPTX) [63]. However, in selected cases, we have undertaken unilateral minimally invasive parathyroidectomy (MIP) with a resulting improvement in the complication profile and acceptable control of HPT [45]. Figure 2 illustrates a case in which unilateral parathyroidectomy has been performed with thymectomy.
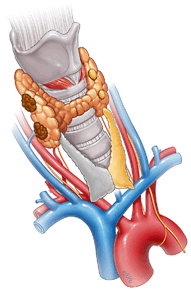
Fig. 2
Minimally invasive parathyroidectomy. In patients with localizing imaging, a focused or unilateral parathyroidectomy approach through minimal incision is recommended. In this procedure, all unilateral glands are explored and resected if found enlarged and at the same time unilateral cervical thymectomy is performed
The technique for total parathyroidectomy involves removing all visible parathyroid glands with autotransplantation of small fragments of parathyroid tissue from the most normal-appearing gland either into the brachioradialis muscle of the forearm or into the sternocleidomastoid muscle of the neck. Once all four parathyroids have been located, cervical thymectomy is undertaken and a portion of the most normal-appearing gland weighing approximately 50 mg is transplanted into the brachioradilais muscle of the nondominant upper limb via a longitudinal incision placed over the forearm [64]. This autotransplantation site is marked with titanium clips to facilitate excision in the event of graft hyperfunction. The remaining glands are removed and sent for histological assessment with a portion of tissue also saved for cryopreservation. The benefits of using cryopreserved tissue in hypoparathyroid patients have also been questioned by other groups who have demonstrated poor success rates after transplantation of cryopreserved tissue [65–69].
sPTX (Fig. 1), in which three and a half glands are removed with a small parathyroid remnant equal to approximately 50 mg left in situ on a vascularized pedicle, is utilized by many especially in the younger patients. Once the viability of the retained gland is established, the remaining glands are removed and the in situ parathyroid is marked with a nonabsorbable Prolene suture. At this point, a cervical thymectomy is performed. Cervical thymectomy has the theoretical advantage of decreasing the rate of MEN-1-associated thymic carcinoid tumors as well as increasing the disease-free survival rates [70–72]. However, it should be noted that cases of thymic carcinoid have been documented in patients with prior cervical thymectomy [73–76].
sPTX is associated with lower rates of hypoparathyroidism on the order of 1–10 % compared with rates of 10–30 % seen in tPTX. However, MEN-1 patients treated with sPTX have been shown to have recurrence rates within 10–12 years between 40 and 60 % of patients, in comparison to sporadic HPT with rates of < 5 % [41, 77–84].
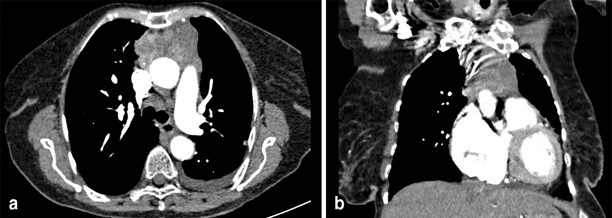
Persistent HPT in MEN-1 is typically due to the failure to identify and resect supernumerary or ectopic glands at the first procedure [85]. In addition, the development of hyperfunction in autotransplanted tissue or parathyroids which were retained at sPTX is a frequent cause of recurrence [85]. Index cases of MEN-1 who undergo surgery for what appears at initial presentation to be sporadic disease are at risk of recurrence as a result of the widespread use of minimally invasive techniques for parathyroidectomy and because hyperplasia may not have been appreciated at the initial procedure. As such, screening patients who present with HPT at a young age, < 40 years for MEN-1, is appropriate. Imaging studies are essential for planning reoperative surgery in contrast to primary cases and having two concordant imaging modalities are ideal prior to proceeding to reoperative parathyroidectomy. Useful modalities include two or more of the following: CT, sestamibi, cervical ultrasonography, MRI with sensitivities of 70–85, 70–85, 25–90, and 60–80 %, respectively [86–90] (Fig. 3). In cases where forearm autotransplantation has been performed, graft hyperfunction can be demonstrated by drawing blood from both arms, while a tourniquet is applied to the side which carries the autotransplanted tissue. Known as the Casanova test, this approach can confirm differential PTH readings between the grafted forearm and peripheral blood and can direct the surgeon to either repeat cervical surgery or removal of the forearm graft. In difficult cases of recurrent disease, where ultrasound, sestamibi, 4-D CT, and MRI are unhelpful, we undertake selective venous sampling (SVS). This modality requires an experienced interventional radiologist and the mapping of PTH gradients from multiple sites along the internal jugular and mediastinal veins. SVS has been shown to correctly localize hyperfunctioning parathyroids in 60–85 % of cases [91–94]. In cases where imaging has failed to confidently localize the site of recurrence, undirected cervical reoperation is usually best avoided, and we prefer to observe the patient for a period of 6 months before repeating our imaging algorithm.
Fig. 3
4-D CT of patient with mediastinal hyperparathyroid. 4-D CT 4-dimensional computerized tomography
After adequate preoperative imaging, reoperation for HPT in MEN-1 can often be undertaken as a focused procedure without the need for extensive dissection. The majority of recurrent intrathymic parathyroid tumors sit within the upper anterior mediastinum and can be successfully removed via cervical exploration. For tumors located deeper in the middle and posterior mediastinum and aorticopulmonary window, minimally invasive thoracic approaches and even formal sternotomy is necessary to establish safe surgical access.
Achieving normocalcemia after revision surgery for HPT in MEN-1 has been described in up to 70 % of cases [72, 95]. Recurrences following reoperation are frequent and occur in 30–60 % of patients with 10–30 % of patients acquiring permanent hypoparathyroidism and 1–5 % with permanent recurrent laryngeal nerve injury. In cases where surgery is not feasible or the patient is not a suitable operative candidate, ethanol ablation and angiographic embolization have been used with some success. Medical therapy with the calcimimetic agent Cinacalcet can be used to manage refractory cases where operative intervention is not appropriate.
Pancreaticoduodenal Neuroendocrine Tumors
Pancreatic and duodenal neuroendocrine tumors (PNETs) are the second most prevalent clinical manifestations of MEN-1 disease with clinical penetrance of 50–70 % [27, 96]. However, autopsy studies have shown that almost 100 % of MEN-1 patients, by the age of 40 years, have multiple nonfunctioning macroadenomas [97]. PNETs can present with clinical syndromes of hormone excess, most commonly from gastrinoma accounting for 30–40 % of cases. However, nonfunctional tumors account for 20 %. Other functioning PNETs include insulinoma (10 %) and rarely glucagonoma, VIPoma, and somatostatinoma (2 %; Table 1) [26].
In contrast to sporadic disease, MEN-1-related PNETs are solid, multifocal, and heterogeneous. Most are microadenomas ( < 0.5m) scattered throughout the pancreas and surround a larger macroadenoma. It has been theorized that these microadenomas may arise from pancreatic duct precursor cells because microadenomas are often surrounded by dysplastic (proliferating) exocrine ducts. Histologically, these lesions do not show normal islet-cell architecture and on immunohistochemistry can stain positively for numerous proteins such as chromogranin A, synaptophysin, neuron-specific enolase, as well as multiple hormones such as PP, glucagon, insulin, and somatostatin [98–101]. In addition, submucosal duodenal carcinoids are detected in > 50 % of patients with MEN-1-associated PNETs which have important implications during surgical intervention [102].
PNETs are the most frequent cause of mortality in MEN-1. At the time of clinical diagnosis, 30–50 % of patients have lymph node or liver metastasis [34, 96, 103, 104]. Furthermore, studies have shown that nearly 50 % of MEN-1-associated deaths before 50 years of age are due to pancreatic malignancy predominantly with liver metastasis [35–37, 104, 105]. Studies looking at the progression of PNETs have shown that they are an important factor in the long-term survival of MEN-1 patients [106, 107]. It remains controversial whether there is a correlation between tumor size and risk of metastasis. However, the French GTE ( Groupe d’etudes des tumeurs endocrines) register did reveal that a tumor size of > 3 cm correlated with a higher rate of metastasis (15–52 %) [105, 108–111]. Interestingly, a recent Swedish study did not show any survival difference between MEN-1 PNET patients compared with similar sporadic cases and that survival itself can be correlated to tumor, node, metastasis (TNM) staging and Ki67 proliferation index [112]. Furthermore, it showed that nonfunctioning tumors had poorer survival. Whether this decreased survival is due to delayed presentation as they are asymptomatic, or that the disease itself is more aggressive, needs to be elucidated.
Functioning Pancreaticoduodenal Endocrine Tumors
Gastrinoma
Zollinger and Ellison in 1955 first described two patients with non-β islet cell pancreatic tumors with marked gastric acid production and subsequent recurrent peptic ulceration [113]. Then in 1960, Gregory was able to extract a gastric secretagogue, otherwise known as Gastrin, from these tumors [114, 115]. As such, gastrinoma is synonymous with the Zollinger–Ellison syndrome. Gastrinoma is a common presentation of MEN-1, and approximately 20–25 % of patients with gastrinoma have MEN-1 disease [116]. The syndrome comprises tumors of the pancreas or duodenum that secrete gastrin and gastrin precursors, thereby mimicking gastrin secreted by G cells in the gastric antrum.
Similarly, to MEN-1 HPT disease, PNETs in these patients differ to sporadic cases, in that age of presentation is usually 10 years younger and that the disease is often multifocal [102, 117]. Gastrinomas in MEN-1 are commonly found in the duodenum ( > 90 %) as small, multiple, and nodular, predominantly in the first and second part which is not dissimilar to sporadic cases [118]. Rarely, gastrinomas can be seen in the pancreas, and these are often large and associated with early liver metastasis.
Studies have shown that many MEN-1 patients have biochemical hypergastrinemia without clinical sequelae of Zollinger–Ellison syndrome. Approximately, 30–50 % of symptomatic patients have nodal or liver metastasis [96, 103, 111, 119]. Those that have metastasized to the liver have a 50 % 5-year survival rate [120]. Despite the multifocal and metastatic potential of gastrinomas in MEN-1, studies have not detected an expected worsened prognosis compared to sporadic cases; 5-year survival rates for all gastrinoma patients range between 62 and 75 % [112]. However, currently there has been no correlation between genotype and disease progression as it can often differ significantly even between family members [34, 111]. Features associated with a worse prognosis are pancreatic primary, liver metastasis, ectopic Cushing’s syndrome, or markedly elevated plasma gastrin concentrations [120].
Localization of gastrinoma may be challenging as these lesions are often small and multifocal. Investigations include CT, MRI, somatostatin receptor scintigraphy, gallium positron emission tomography (Ga-PET) scan and endoscopic ultrasound. CT and MRI are important in evaluating the pancreas, liver, lymph node status, and overt peritoneal disease. Figure 4 is an example of a CT scan of a patient with PNETs. Endoscopic ultrasound also has high sensitivity of up to 90 % and has the added advantage of incorporating fine-needle biopsy of the suspected lesions [121–123]. Endoscopic ultrasound and intraoperative ultrasound have become the most reliable modalities for isolating pancreatic and duodenal lesions in our experience. Fluorodeoxyglucose (FDG) PET scan has a limited role in detecting most primary PNETs as they are commonly negative due to lesions being less than 5 mm. However, FDG PET is useful for evaluating metastatic disease. The recent use of Gallium 68-labeled octreotide scintigraphy (Ga-68 1,4,7,10-tetraazacyclododecane-NI,NII,NIII,NIIII-tetraacetic acid (D)-Phe1-thy3-octreotide (DOTATOC)) allows functional detection of the lesions and can often identify up to 35 % of occult tumors not seen in anatomical imaging [124]. It has a sensitivity of around 85 % and specificity of up to 100 % [124–126].
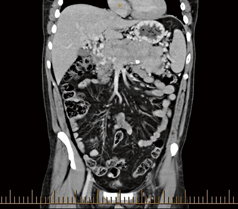
Fig. 4
CT scan in coronal view demonstrating enteropancreatic endocrine tumor. As can be seen the multifocal nature of the pancreatic lesions can be appreciated. CT computerized tomography
Treatment of gastrinoma involves managing the hypergastrinemia and surveillance for the development of malignancy. Medical treatment with acid inhibitors, such as proton pump inhibitors (PPIs), has markedly reduced the rate of death resulting from hypergastrinemia-induced metabolic complications. However, medical treatment does not prevent malignant transformation or metastatic disease, and this is where surgical intervention may play a role.
The aim of surgical intervention is to provide eugastrinemia as well as removing malignant or premalignant disease. There are several controversies regarding surgical management of gastrinoma in MEN-1, and these include timing and indication of surgery, type of surgery, and surgery for advanced disease. PNETs in MEN-1 are multifocal and as such curative excision of gastrinomas is unlikely. Norton, in 1999, in a prospective study showed that cure rates were 5 % at 5 years and 0 % at 10 years [118]. In comparison, sporadic cases had a cure rate of 34 % at 10 years. Furthermore, postoperative analysis demonstrated no survival benefit or decrease in rate of liver metastasis in patients who had undergone surgical excision [118, 127]. However, it should be noted in these studies, indications for surgery were lesions greater than 3 cm and/or symptoms of obstruction. Proponents for delayed surgical intervention argue that even with metastatic disease, there is a 15-year survival rate of 52 % [128]. Lowney, in a recent study of MEN-1 patients, could not demonstrate a correlation between size of tumor and metastatic potential in general, only those with tumors of >survival benefit in the form of reducing liver metastasis. Studies 2 cm had liver metastasis [105]. This raises the question whether earlier surgical intervention may actually produce survival benefit in the form of reducing liver metastasis. Studies in support of this idea involved resection of lesions > 1 cm and have shown no development of liver metastasis after a median follow-up of 83 months [111]. Finally, it has been suggested that there should be a delay in operating in small PNETs because surgery itself has a mortality rate of up to 15 %. However, it should be noted there have been numerous studies with little or no mortality associated with surgical intervention [111, 129–131].
There have been many surgical approaches advocated in the literature, and the most commonly described is that of Dr. Norman Thompson. This surgical approach is primarily based on two unique characteristics of PNET. The first is that gastrinomas are usually located in the duodenum and within the “gastrinoma triangle” between the junction of the cystic and common bile ducts, junction of second and third parts of duodenum, and junction of the neck and body of the pancreas. Second, concomitant nonfunctioning tumors are usually found in patients with gastrinoma. Consequently, Thompson proposed that intraoperative ultrasound should be used to investigate lesions in the pancreas, and those found in the pancreatic head are enucleated. At the same time, an 80 % distal pancreatectomy with splenic preservation is performed (Fig. 5). Those with Zollinger–Ellison syndrome are further subjected to duodenotomy, whereby the entire duodenum is examined and the typical small submucosal lesions are enucleated. A regional lymphadenectomy is also undertaken. Initial results reported by Thompson using this technique in 40 patients have demonstrated that 68 % of patients remain eugastrinemic and that only one person developed a solitary liver metastasis with no mortality in follow-up as long as 19 years [121, 122]. However, long-term follow-up of these patients have demonstrated a high recurrence rate of PNETs in the remaining pancreas [132]. There is a 10–20 % risk of pancreatic pseudocyst and fistula which are often dealt with adequately with drainage and time. Controversy exists whether routine distal pancreatectomy should be performed. The theoretical advantage of distal pancreatectomy in patients without evidence of distal pancreatic disease has been questioned with studies like that of Norton et al. showing excellent survival rates without routinely performing distal pancreatectomy. In their study of 81 patients, 25 of these with PNET < 2.5 cm had 15-year survival of 100 %, 17 patients with PNET < 6 cm had survival of 100 %, and 31 patients with ³ 2 tumors or > 6 cm had survival of 89 % [106, 133].
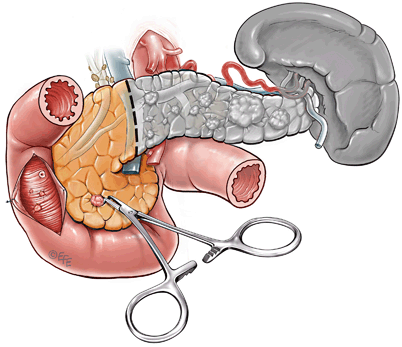
Fig. 5
Surgical management of large distal pancreatic lesions. This includes distal pancreatectomy (80 %) with enucleation of smaller lesions in pancreatic head. Patients who have a raised gastrin level will then proceed to have a duodenotomy to allow identification and enucleation of duodenal gastrinomas
For MEN-1 cases with larger lesions in the head of the pancreas which are not amenable to conservative enucleation, pylorus preserving pancreaticoduodenectomy (PPPD) is required (Fig. 6). Rarely, patients with multiple large malignant pancreatic lesions may require total pancreatectomy; however, these cases are almost always associated with significant endocrine and exocrine insufficiency, have higher complication rates, and should be reserved for those kindreds where a strong history of metastatic disease is apparent [131, 134].
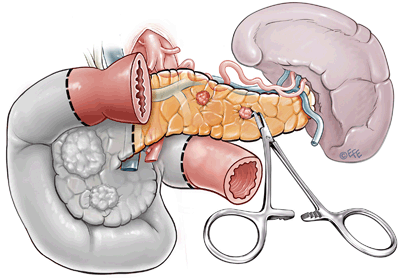
Fig. 6
Surgical management of large lesions in pancreatic head. This involves pylorus-preserving pancreaticoduodenectomy with enucleation of distal pancreatic lesion
Patients with recurrent disease can often undergo reoperation with either further enucleations or pancreaticoduodenectomy. A study by Jaskowiak exploring reoperations in patients with recurrent disease found that the 2.5-year follow-up cure rate was lower at 23 % (vs. 47 % in those with only a primary operation) [135]. The authors suggested that cure rates of 23 % were a clear indication that surgery should be attempted to allow patients to be “disease free.” However, 15-year survival rates of 93 % are seen in post-operative patients and as such reoperation may not change survival [133]. Furthermore, the role of surgical intervention in hepatic metastasis is evolving. Surgical debulking and/or radiofrequency ablation of hepatic metastasis with acceptable morbidity and low mortality is generally recommended as it may increase survival as well as help manage Zollinger–Ellison syndrome. For patients with disseminated disease, hormonal control with long-acting octreotide, PPIs, chemotherapy agents, such as streptozotocin and 5-fluorouracil or doxorubicin, have been used with some success for palliation [136].
Insulinoma
Insulinoma are β islet cell tumors that secrete insulin and are the second most common functional PNETs seen in MEN-1 patients with a prevalence of 10 % (Table 1) [27, 29]. MEN-1-associated insulinoma accounts for only 4 % of all patients with insulinoma. Patients are younger at presentation (less than 20 years of age) which is in contrast to those sporadic cases which typically present greater than 40 years of age. Clinically, they present with hormonal effects of hyperinsulinemia, hypoglycemia, especially after fasting or exertion. Diagnosis requires supervised 72-h fasting with inappropriately elevated insulin C-peptide and proinsulin concentrations. MEN-1 insulinoma is usually caused by a single dominant and benign tumor and is typically 2–3 cm in size but can be located anywhere in the pancreas [29, 137]. However, it is often associated with multiple other nonfunctioning lesions. Surgical excision remains the most effective treatment for insulinoma in order to prevent life-threatening hypoglycemia.
Preoperative localization studies include endoscopic ultrasound, CT, celiac axis angiography, pre- and perioperative transhepatic portal venous sampling, selective intra-arterial stimulations with hepatic venous sampling, and intraoperative direct pancreatic ultrasonography. Operative management is dependent upon the site, size, and proximity to the pancreatic duct of the dominant lesion. Cure rates are greater than 90 % with median follow up of 10 years and conservative resections as expected have a higher incidence of recurrence versus subtotal resections [138–140].
VIPoma, glucagonoma, PPoma, Somatostatinoma, and GHRHoma
Pancreatic tumors which secrete vasoactive intestinal peptide (VIP), the so-called Verner–Morrison syndrome, glucagon, PP, somatostatin, and growth-hormone-releasing hormone (GHRH) occur infrequently. These are all rare and together have a prevalence of less than 2 % in MEN-1 syndrome [27, 141]. In PPoma and somatostatinoma, secretion of these enzymes usually does not translate into any apparent endocrinopathy. In contrast, tumors that secrete growth-hormone-releasing hormone (GHRH) have rarely been reported in MEN-1 and when they arise can be found > 50 % in the lung, 30 % in pancreas, and 10 % in small intestine. Glucagonoma usually presents as hyperglycemia, anorexia, glossitis, anemia, diarrhea, venous thrombosis and necrolytic migratory erythema. By the time patients present, the lesions are large and have often metastasized and palliative surgery, and ablative procedures are typically necessary.
VIPomas secrete VIPs causing watery diarrhea, hypokalemia, and achlorhydria. As these lesions are usually located in the tail of the pancreas, surgical excision has been curative. Unfortunately, unlike insulinomas, these rarer PNETs are often asymptomatic until the tumor is large and metastasis to visceral organs is present. The GTE registry showed that these tumors were twice as likely to be > 3 cm in size and visceral metastasis were also more frequently seen in comparison to other types of MEN-1-associated PNETs (40 and 15 %, respectively). These characteristics translate to a poorer survival that is equivalent to that of nonfunctioning tumors. The 10-year disease-related survival rates for the various PNETS is as follows: glucagonomas, VIPomas, or somatostatinomas 54 %; gastrinomas 81.7 %; insulinomas 91.4 %; and nonfunctioning PNET 62.2 % (141).
Nonfunctioning Pancreaticoduodenal Endocrine Tumors
The prevalence of nonfunctioning PNETs in MEN-1 is around 20–35 % and clinical presentation has been detected as early as 12 and 14 years of age [26, 27, 108]. Despite its asymptomatic nature, they have a high-malignant potential and subsequently need to be monitored and resected before malignant progression. The 10-year survival rate for patients with nonfunctioning PNETs has been found to be around 62 % [108, 141]. Furthermore, the rate of survival can be correlated to tumor size and metastasis. Tumors with diameters of < 1, 1–3, and > 3 cm had metastasized in 4, 28, and 43 % of patients, respectively. This correlated with 8-year survival rates of approximately 100, 80, and 40 %, respectively [108]. Studies by Triponez and colleagues have recommended surgical excision in those that are ≥ 2 cm. However, in their study, 4 % of patients with tumors, less than 1 cm had metastasis [108, 109]. Due to this, some have called for earlier intervention on lesions > 1 cm. Whether preventing metastasis by more aggressive and earlier surgical intervention is improving survival has yet to be elucidated.
Pituitary Tumors
Anterior pituitary tumors are found in about 30 % of MEN-1 patients and account for 25 % of its initial presentation [26, 27]. Among all pituitary adenomas which proceed to surgical resection, 2–3 % occurs in the setting of MEN-1. In the French GTE registry, out of 324 patients, 42 % presented with pituitary adenomas with mean age of onset of 38 years but ranged from 12 to 83 years of age [108, 110]. The distribution of the types of pituitary adenoma is similar to sporadic cases; prolactinoma in 62 %, somatotropinoma in 9 %, Cushing’s disease in 4 %, and nonsecreting in 15 % [110, 142]. MEN-1-associated pituitary adenomas were generally seen to be larger, and more commonly invasive than sporadic cases. In addition, there have been reported cases of double adenomas and multi-hormonal associations such as prolactin and adrenocorticotropic hormone (ACTH) or prolactin/growth hormone (GH) and follicle stimulating hormone/luteinizing hormone (FSH/LH) which are all atypical [143]. Clinical manifestations and treatment are similar to those with sporadic pituitary tumors. Surgery, medical therapy with dopamine agonists, and radiotherapy have all been used. The preferred treatment depends on the tumor size and invasion, visual impairment, the presence of hormonal hypersecretion, and response to medical treatment.
Other MEN-1-Associated Tumors
Carcinoid Tumors
Carcinoid tumors in MEN-1 patients are found predominately in the foregut (gastric, thymic, and bronchial). Carcinoids occur in about 15 % of MEN-1 patients [26, 27, 29]. MEN-1 carcinoid tumors also have strong gender-specific distribution; thymic carcinoids are found predominantly in males and bronchial carcinoids in females [24, 71, 144, 145]. Furthermore, unlike other MEN-1 associated tumors, the average age of onset is much later in the 40s [146]. This reflects the lack of hormonal oversecretion and compression-induced symptoms. These tumors have malignant potential and represent the second most common case of tumor-related deaths. Thymic carcinoids are more often malignant than bronchial carcinoids (70 vs. 20 %, respectively).
Thymic carcinoids are usually found at an advanced stage as a large invasive mass as can be seen in Fig. 7. Patients with MEN-1 thymic carcinoid have a poor prognosis with reported median survival of 28 months and 5-year survival rate of less than 30 %. As such, it has been suggested that routine transcervical thymectomy should be performed as part of procedures for parathyroid disease [70–72]. However, resection does not prevent development of thymic carcinoids as there have been many reported cases of post-cervical thymectomized patients with thymic carcinoids [73–76]. It seems rational therefore to carefully monitor for progression of thymic disease. Bronchial carcinoids on the other hand are more indolent. Gastric carcinoids are tumors of enterochromaffin-like cells, and they occur in 10 % of MEN-1 patients. They can be found either as an incidental finding on endoscopy or cause hormonal sequelae by its excessive secretion of serotonin and histamine. Also known as type II gastric neuroendocrine tumours, they have a 5-year disease free survival rate of 78%. Lesions smaller than 2 cm are recommended for endoscopic removal; however, larger ones require surgical excision.