Typical manifestations
MEN 2 subtypes
MEN 2A (%)
MEN 2B (%)
FMTC (%)
Medullary thyroid cancer
100
100
95–100
Pheochromocytoma
50
50
–
Primary hyperparathyroidism
15–30
–
–
Gangliotomosis of GI tract
–
100
–
Mucosal neuromas
–
100
–
Marfanoid habitus
–
75
–
MEN 2A
MEN 2A is also known as Sipple syndrome named after John Sipple, a pulmonary physician who first reported this endocrinopathy [1, 2]. This syndrome includes MTC , pheochromocytoma, and primary hyperparathyroidism. MTC is the most common clinical presentation of MEN 2A, with nearly 100 % of patients affected. The average age of presentation of a patient with sporadic MTC is in the sixth decade. In contrast, the average age of presentation for a patient with MEN 2A syndrome is 20–40 years, with some more aggressive forms presenting by age 5 [6].
Less commonly, patients present with symptoms due to pheochromocytomas. Pheochromocytomas are present in approximately 50 % of patients with MEN 2A and cause various symptoms including palpitations, headaches, hypertension, and flushing. Hyperparathyroidism occurs in 15–30 % of MEN 2A patients, with the peak incidence in the third or fourth decade. Multiglandular hyperplasia is the most common histologic finding, although single gland adenomas are present in some patients [4, 5, 7]. In addition, rare variants of MEN 2A exist, which include the above-listed endocrine tumors, along with either cutaneous lichen amyloidosis or Hirschsprung’s disease [8, 9].
MEN 2B
Like MEN 2A, MEN 2B patients have MTC and pheochromocytomas, but they do not present with parathyroid disease. Instead, MEN 2B patients exhibit various unique physical features, most commonly mucosal neuromas and a marfan-like physical constitution.
MTC in MEN 2B also manifests in nearly 100 % of patients. MTC develops earlier and more aggressively than in MEN 2A. Metastatic disease has been reported before age 1, and death may occur in the second or third decade of life [4, 5]. Pheochromocytomas occur in more than half of MEN 2B patients with similar presentations as in MEN 2A. There are no well-documented examples of parathyroid disease in MEN 2B patients.
The mucosal neuromas and marfanoid body habitus are the most distinctive features of MEN 2B and are recognizable in childhood. Patients can have prominent lips and prognathic jaws (Fig. 1). The corneal nerves can also be quite enlarged. The marfanoid habitus combined with facial features is unique, and often unrelated affected individuals look strikingly similar. Neuromas are present on the tip of the tongue, buccal mucosa, under the eyelids, and throughout the gastrointestinal (GI) tract. The most common presentation in children relates to GI symptomatology, including intermittent colic, pseudo-obstruction, and diarrhea. Some children present with failure to thrive secondary to these persistent GI symptoms [5, 10]. Almost 75 % of patients will have skeletal abnormalities that give them a marfanoid body habitus [11]. Mucosal neuromas or GI gangliomas are present in nearly 100 % of MEN 2B patients [12]. Confirmation of these various clinical features can be made by biopsy of the mucosal neuromas, rectal biopsy, or slit-lamp examination for medullated corneal nerves [6].
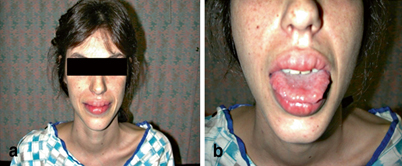
Fig. 1
Some distinct physical characteristics of MEN 2B patients. a MEN 2B patient with typical thin, marfanoid habitus, prognathic jaws, and prominent lips. b Mucosal neuromas are often found on the lips and tongue. (Courtesy of Quan-Yang Duh, UCSF, Department of Surgery)
FMTC
Once thought to be a distinct entity, first described by John Farndon, FMTC is now considered a common subvariant of MEN 2A, in which MTC is the only manifestation. The clinical diagnosis of FMTC is established by the presence of MTC in greater than four family members over multiple generations without any evidence of pheochromocytoma or hyperparathyroidism. The clinical course of MTC in FMTC patients is more benign than in MEN2A and MEN2B, with a late onset or no clinical manifestation of disease. Thus, the overall prognosis is relatively good and better than the other MEN 2 subtypes [8, 9].
Since the penetrance of pheochromocytoma is 50 % in MEN 2A, it is possible that MEN 2A could masquerade as FMTC in small kindreds. It is important to consider this, because failure to recognize this scenario could lead to serious morbidity and mortality from an undiagnosed pheochromocytoma in an affected family member [4, 5].
Medullary Thyroid Cancer
Up to 75 % of all MTCs are thought to be sporadic; the rest are hereditary in the form of MEN 2 syndromes [5]. MTC is present in nearly all patients with MEN 2A and MEN 2B and is often the first presenting disease of the syndrome. The calcitonin-producing cells (C-cells) of the thyroid, which are derived from the neural crest, are considered to be the precursor cells from which MTC arises. This tumor usually develops in childhood, beginning as hyperplasia of the C-cells [5]. The C-cells secrete calcitonin and carcinoembryonic antigen (CEA), which serve as tumor markers for the disease. While sporadic MTC can present as a solitary nodule, hereditary MTC tends to be multicentric. The disease spreads regionally from central to lateral neck lymph nodes and then to the liver, lung, bone, and brain [8].
Initially, biochemical testing with basal and stimulated levels of calcitonin was used as the primary screening method for diagnosis of hereditary MTC. Secretion of calcitonin can be stimulated 3–20-fold with intravenous (IV) pentagastrin, calcium, or both [7]. The effectiveness of pentagastrin-stimulated calcitonin screening increases progressively with age, though, unfortunately, lacks sensitivity where it is needed most—in young children. Thus, its use as screening for MEN 2 syndrome in children before the age of 5 has its limitations [4, 6]. More recently, advances in genetic analysis of the RET oncogene mutations has led to direct DNA analysis becoming the primary screening method for MTC. Calcitonin screening is still used by some to determine the timing of prophylactic thyroidectomy for patients with certain RET mutations [8]. After surgical resection, plasma calcitonin and CEA levels are used to monitor disease progression. Normalization of these tumor markers has a favorable prognosis, whereas progressive increases in calcitonin or CEA indicate persistent or recurrent disease [8, 9, 13]. Calcitonin and CEA tend to rise together, but in some cases of recurrent disease, calcitonin will no longer be elevated, while CEA levels are. This is considered a sign of tumor dedifferentiation [4, 8].
Due to the poor prognosis of advanced stage MTC, early prophylactic surgery is recommended before the disease is clinically evident or becomes regionally advanced. For patients with MEN 2A and FMTC under the age of 5 with no clinically evident lymph nodes, prophylactic total thyroidectomy alone is the recommended operation. For both node-negative MEN 2A and FMTC patients older than 8 years and all MEN 2B patients, prophylactic total thyroidectomy with central neck (level VI) dissection is indicated. In newly diagnosed adult MEN 2 patients with MTC, total thyroidectomy should be performed with node dissection based on extent of disease. In patients with no evidence of local invasion or local cervical metastases, total thyroidectomy and prophylactic central neck lymph node dissection is advocated. In patients with limited locally advanced disease, lateral neck dissection is also supported [4]. Recommendations for timing of surgical resection based on genotype are discussed later in the chapter.
Pheochromocytoma
Pheochromocytomas are present in about 50 % of both MEN 2A and MEN 2B patients. The characteristics of the tumors and clinical manifestations are similar in the two subvariants. Pheochromocytomas arise in the adrenal medulla from neural crest-derived chromaffin cells which primarily secrete the catecholamines epinephrine and norepinephrine. Tumors tend to be benign with metastasis being uncommon. Tumors are commonly bilateral. If they do not present simultaneously, more than 50 % of patients who have had unilateral adrenalectomy later develop a pheochromocytoma in the contralateral gland within a decade. A distinguishing feature from hereditary paraganglioma syndromes is that the pheochromocytomas in MEN 2 are almost always found within the adrenal gland, not as extra-adrenal tumors [4, 5, 7]. Clinical presentation is similar to sporadic forms with patients having symptoms from catecholamine excess such as headaches, palpitations, hypertension, tachycardia, and flushing.
Screening for pheochromocytoma involves measuring plasma or 24-h urinary levels of metanephrines and normetanephrines. If the biochemical diagnosis is confirmed, the patient should get imaging to localize the tumor with either computed tomography (CT) or magnetic resonance imaging (MRI) [4]. Further localization and staging for metastatic disease is performed with radiolabeled metaiodobenzylguanidine (MIBG) scintigraphy and positron emission tomography (PET) with different radiotracers such as fluorine-18-dopamine ([18F]DA), fluorine-18-dihydroxyphenylalanine ([18F]DOPA) or fluorine-18-fluorodeoxyglucose ([18F]FDG). MIBG scans have historically been the most widespread functional imaging technique used for localization of pheochromocytomas and paragangliomas with high sensitivity and specificity [14, 15]. However, recent studies have shown that MIBG has lower sensitivity for certain conditions such as familial pheochromocytoma and paraganglioma syndromes and malignant disease. Increasing evidence suggests that fluorine dopamine (FDA)-PET, fluorodeoxyglucose (FDG)-PET, and dihydroxyphenylalanine (DOPA)-PET are superior localization studies for pheochromocytoma and are helpful in the above scenarios when MIBG fails to demonstrate disease, though suspicion for disease is high (Fig. 2). MIBG is still the most widely used functional imaging technique and has the added advantage of selecting for patients with metastatic disease who are suitable for MIBG nuclear therapy [14, 15].
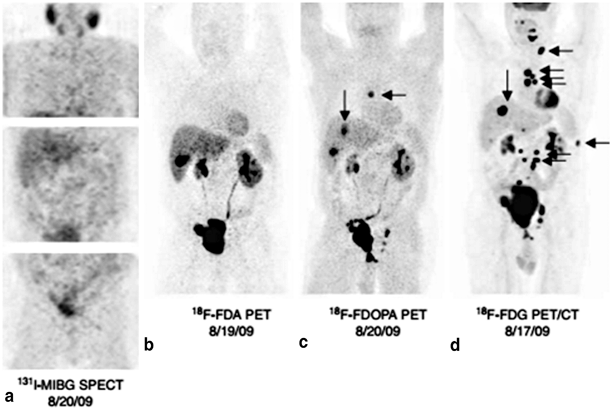
Fig. 2
Comparison of detection of metastatic pheochromocytoma by various nuclear imaging techniques. Nuclear imaging studies of a 55-year-old male with metastatic pheochromocytoma who tested negative for succinate dehydrogenase B, C, and D mutations. His primary tumor was located in the bladder and was initially resected in 2005. In 2009, he presented with metastatic disease. His 131 I-MIBG SPECT was negative (a), but his other nuclear imaging studies show metastatic disease: [18F]-FDA PET (b), [18F]-FDOPA PET (c), and especially [18F]-FDG PET/CT (d). (From [16]. Reprinted with permission from Bioscientifica, Ltd)
Because of the possible coexistence of pheochromocytoma, it is essential to screen for these tumors before thyroidectomy for MTC. There is significant morbidity and mortality from an undiagnosed pheochromocytoma. Before the association of pheochromocytomas and MTC in MEN 2 was recognized, many patients died from a hypertensive crisis during or after thyroidectomy secondary to unrecognized disease [1]. In fact, before advances in screening, pheochromocytoma, not MTC, was the leading cause of death in MEN 2A patients. If a patient has both a pheochromocytoma and MTC, adrenalectomy should be performed first to avoid an intraoperative adrenal crisis during thyroidectomy [4].
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


