Abbreviations
- 11β-HSD2 11β-Hydroxysteroid dehydrogenase type 2
- ACE Angiotensin-converting enzyme
- ACTH Corticotropin
- AME Apparent mineralocorticoid excess
- ANP Atrial natriuretic peptide
- APA Aldosterone-producing adenoma
- ARB Angiotensin receptor blocker
- AVS Adrenal venous sampling
- CAH Congenital adrenal hyperplasia
- CT Computed tomography
- DOC Deoxycorticosterone
- FH Familial hyperaldosteronism
- GH Growth hormone
- GRA Glucocorticoid-remediable aldosteronism
- HU Hounsfield units
- IHA Idiopathic hyperaldosteronism
- IVC Inferior vena cava
- PAC Plasma aldosterone concentration
- PAH Primary adrenal hyperplasia
- PRA Plasma renin activity
Endocrine Hypertension: Introduction
Hypertension affects one in four adults in the developed world. Although hypertension is essential or idiopathic in most cases, a cause can be detected in approximately 15% of the hypertensive population. The secondary causes of hypertension can be divided into renal (eg, renal vascular or parenchymal disease) and endocrine causes. There are at least 14 endocrine disorders in which hypertension may be the initial clinical presentation (Table 10–1). The diagnosis of endocrine hypertension presents the clinician an opportunity to provide a surgical cure or to achieve a marked response with targeted pharmacologic therapy. Pheochromocytoma and Cushing syndrome are reviewed in detail in Chapters 11 and 9, respectively. The renin-angiotensin-aldosterone system, the diagnostic and therapeutic approaches to mineralocorticoid hypertension (eg, primary aldosteronism), and less common forms of endocrine hypertension are reviewed in this chapter.
Adrenal Dependent Pheochromocytoma Primary aldosteronism Hyperdeoxycorticosteronism Congenital adrenal hyperplasia 11β-Hydroxylase deficiency 17α-Hydroxylase deficiency Deoxycorticosterone-producing tumor Primary cortisol resistance Cushing syndrome Apparent Mineralocorticoid Excess (AME)/11β-Hydroxysteroid Dehydrogenase Deficiency Genetic Type 1 AME Acquired Licorice or carbenoxolone ingestion (type 1 AME) Cushing syndrome (type 2 AME) Thyroid Dependent Hypothyroidism Hyperthyroidism Pituitary Dependent Acromegaly Cushing disease |
Renin-Angiotensin-Aldosterone System
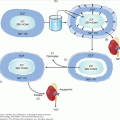
The components of the renin-angiotensin-aldosterone system are shown in Figure 10–1. Aldosterone is secreted from the zona glomerulosa under the primary control of angiotensin II, potassium, and corticotropin (ACTH). The secretion of aldosterone is restricted to the zona glomerulosa because of zonal-specific expression of aldosterone synthase (CYP11B2). Atrial natriuretic peptide (ANP), dopamine, and heparin inhibit aldosterone secretion.
Figure 10–1
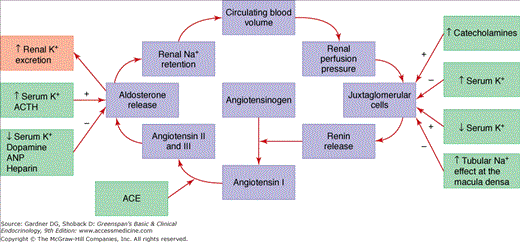
Renin-angiotensin-aldosterone and potassium-aldosterone feedback loops. Zona glomerulosa aldosterone production and secretion are determined by input from each loop (ACE, angiotensin-converting enzyme; ACTH, cortocotropin; ANP, atrial natriuretic peptide; BP, blood pressure; K+, potassium; Na+, sodium).
Renin is an enzyme produced in the juxtaglomerular apparatus of the kidney, stored in granules, and released in response to specific secretagogues. The first 43 amino acids of the 340 amino acid renin protein are a prosegment cleaved to produce the active enzyme. The release of renin into the circulation is the rate-limiting step in the activation of the renin-angiotensin-aldosterone system. Renal renin release is controlled by juxtaglomerular cells acting as pressure transducers that sense stretch of the afferent arteriolar wall and thus renal perfusion pressure; the macula densa, a specialized group of convoluted distal tubular cells that function as chemoreceptors for monitoring the sodium and chloride loads present in the distal tubule; the sympathetic nervous system, which modifies the release of renin, particularly in response to upright posture; and humoral factors, including potassium, angiotensin II, and ANPs. Thus, renin release is maximized in conditions of low renal perfusion pressure or low tubular sodium content (eg, renal artery stenosis, hemorrhage, dehydration). Renin release is suppressed by elevated perfusion pressure at the kidney (eg, hypertension) and high sodium diets. Renin release is increased directly by hypokalemia and decreased by hyperkalemia.
Angiotensinogen, an α2-globulin synthesized in the liver, is the substrate for renin and is broken down into the angiotensin peptides. Angiotensinogen consists of 485 amino acids, 33 of which constitute a signal peptide that is cleaved prior to secretion. The action of renin on angiotensinogen produces angiotensin I. Angiotensin I is composed of the first 10 amino acid sequence following the signal peptide and does not have biologic activity. Angiotensin II, the main form of biologically active angiotensin, is formed by cleavage of the two carboxyl-terminal amino acids of angiotensin I by angiotensin-converting enzyme (ACE) (Figure 10–2). ACE is localized to cell membranes in the lung and intracellular granules in certain tissues that produce angiotensin II. Amino peptidase A removes the amino-terminal aspartic acid to produce the heptapeptide, angiotensin III. Angiotensin II and angiotensin III have equivalent efficacy in promoting aldosterone secretion and modifying renal blood flow. The half-life in the circulation of angiotensin II is short (< 60 seconds). Elements of the renin-angiotensin-aldosterone system are present in the adrenal glands, the kidneys, the heart, and the brain. For example, the adrenal glomerulosa cells contain the proteins needed to produce and secrete angiotensin II. Other tissues contain one or more components of the renin-angiotensin system and require other cells or circulating components, or both, to generate angiotensin II.
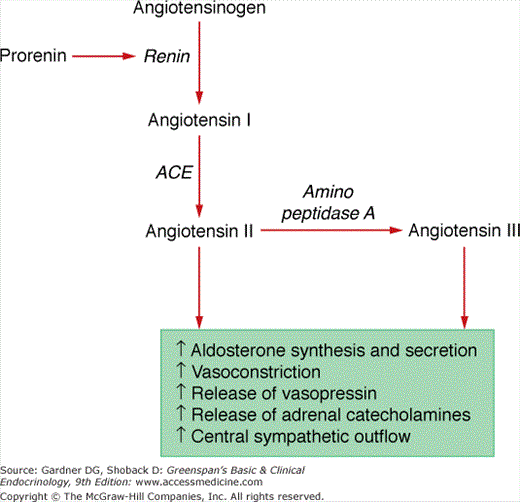
Angiotensin II functions through the angiotensin receptor to maintain normal extracellular volume and blood pressure by (a) increasing aldosterone secretion from the zona glomerulosa by increasing transcription of CYP11B2; (b) constriction of vascular smooth muscle, thereby increasing blood pressure and reducing renal blood flow; (c) enhancing the release of norepinephrine and epinephrine from the adrenal medulla; (d) enhancement of the activity of the sympathetic nervous system by increasing central sympathetic outflow, thereby increasing norepinephrine discharge from sympathetic nerve terminals; and (e) promotion of the release of vasopressin.
Aldosterone is produced in the zona glomerulosa of the adrenal cortex. Approximately 50% to 70% of aldosterone circulates bound to either albumin or corticosteroid-binding globulin; 30% to 50% of total plasma aldosterone is free. Aldosterone is rapidly inactivated to tetrahydroaldosterone in the liver and has a half-life of 15 to 20 minutes. Aldosterone regulates extracellular volume and potassium homeostasis by binding to renal cortical collecting duct principal epithelial cell mineralocorticoid receptors (Figure 10–3). The mineralocorticoid receptor—a member of the nuclear receptor family and also found in the heart, colon, and hippocampus—is localized to the cytoplasm prior to activation, undergoes a conformation change on binding to aldosterone, and translocates into the nucleus where it functions as a transcription factor. The aldosterone-regulated serum- and glucocorticoid-inducible kinase appears to be a key intermediary (see Figure 10–3). Aldosterone increases expression of this kinase which phosphorylates and inactivates neural-precursor-cell-expressed, developmentally down regulated (Nedd) 4-2, a ubiquitin ligase which is responsible for degrading the epithelial sodium channel. This, in turn, leads to an increased number of open sodium channels in the luminal membrane of the principal cells in the cortical collecting tubule, resulting in increased sodium ion reabsorption. The sodium loss increases luminal electronegativity, which augments tubular secretion of potassium by the renal tubular cells and hydrogen ion by the renal interstitial cells. Another mediator of the mineralocorticoid receptor transcriptional response is the activation is the sodium-potassium ATPase at the basolateral membrane, which drives the uptake of potassium and export of sodium (see Figure 10–3). Although glucocorticoids and mineralocorticoids bind equally to the mineralocorticoid receptor, specificity of action is due to the glucocorticoid-degrading enzyme, 11β-hydroxysteroid dehydrogenase, which is strongly expressed in the kidney and prevents glucocorticoids from interacting with the receptor.
Figure 10–3
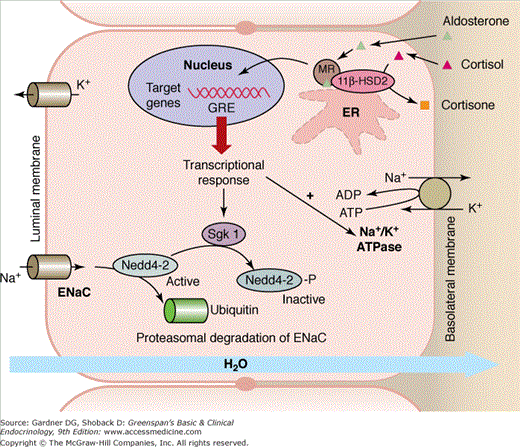
Aldosterone regulates extracellular volume and potassium homeostasis by binding to the renal cortical collecting duct principal epithelial cell mineralocorticoid receptor (MR). The activated MR translocates into the nucleus where it binds to the glucocorticoid response element (GRE) and functions as a transcription factor. Aldosterone increases expression of serum- and glucocorticoid-inducible kinase (Sgk1), which phosphorylates and inactivates neural-precursor-cell-expressed, developmentally downregulated gene (Nedd) 4-2, a ubiquitin ligase which is responsible for degrading the epithelial sodium channel (ENaC). Another mediator of mineralocorticoid receptor transcriptional response is the activation is the sodium-potassium ATPase (Na+/K+ ATPase) at the basolateral membrane, which drives the uptake of potassium and export of sodium. Although glucocorticoids and mineralocorticoids bind equally to the mineralocorticoid receptor, specificity of action is due to the glucocorticoid-degrading enzyme, 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), which prevents glucocorticoids from interacting with the receptor. (Adapted from Odermatt A, Atanasov AG. Mineralocorticoid receptors: Emerging complexity and functional diversity. Steroids 2009; 74:163-171.)
Aldosterone has nonclassic effects that, although probably genomic and therefore mediated by activation of the cytosolic mineralocorticoid receptor, do not include modification of sodium-potassium balance. Aldosterone-mediated actions include the expression of several collagen genes; activation of genes controlling tissue growth factors, such as transforming growth factor β and plasminogen activator inhibitor type 1; or increased expression of genes mediating inflammation. The resultant actions lead to microangiopathy, necrosis (acutely), and fibrosis in various tissues such as the heart, the vasculature, and the kidney. Increased levels of aldosterone are not necessary to cause this damage; an imbalance between the volume or sodium balance state and the level of aldosterone appear to be the critical factors. Spironolactone and eplerenone are mineralocorticoid receptor antagonists. Mineralocorticoid receptor blockade has proven to be clinically important in patients with cardiovascular disease. For example, when spironolactone was added to the treatment program for patients with New York Heart Association class IV heart failure or class III heart failure, it resulted in a significant 30% reduction in overall mortality due to reductions in death from heart failure and sudden death. When eplerenone was added to the treatment program for patients who had a myocardial infarction 3 to 14 days previously and had a left ventricular ejection fraction of ≤40 percent, it resulted in a significantly lower rate of cardiovascular mortality and sudden cardiac death. The effect of mineralocorticoid receptor antagonists on survival in patients with primary aldosteronism has not yet been studied.
The action of angiotensin II on aldosterone synthesis and secretion involves a feedback loop that also includes extracellular fluid volume (see Figure 10–1). A decrease in circulating blood volume results in decreased renal perfusion pressure that is detected by the renal juxtaglomerular cells. Activation of the juxtaglomerular cells increases renin release, which catalyzes the conversion of angiotensinogen to angiotensin I. Angiotensin-converting enzyme (ACE) in the pulmonary and renal endothelium catalyzes the conversion of angiotensin I to angiotensin II and III, which act on the adrenal zona glomerulosa angiotensin receptor to stimulate aldosterone release. Aldosterone acts at the renal mineralocorticoid receptors to stimulate sodium and water retention to preserve the circulating blood volume. Renin release can also be triggered by catecholamines, hypokalemia, and a decrease in sodium chloride absorption in the macula densa cells. Aldosterone secretion can be directly stimulated by ACTH and hyperkalemia. Thus, sodium restriction activates and sodium overload suppresses the renin-angiotensin-aldosterone axis. Mineralocorticoid escape refers to the counterregulatory mechanisms that occur after 3 to 5 days of excessive mineralocorticoid administration. Several mechanisms contribute to this escape, including renal hemodynamic factors and increased release of ANP.
Excess aldosterone secretion causes hypertension through two main mechanisms: (1) mineralocorticoid-induced expansion of plasma and extracellular fluid volume; and (2) increase in total peripheral vascular resistance.
Primary Aldosteronism
Hypertension, suppressed plasma renin activity (PRA), and increased aldosterone excretion characterize the syndrome of primary aldosteronism. Aldosterone-producing adenoma (APA) and bilateral idiopathic hyperaldosteronism (IHA) are the two most common subtypes of primary aldosteronism (Table 10–2). A much less common form, unilateral hyperplasia, is caused by micronodular or macronodular hyperplasia of the zona glomerulosa of predominantly one adrenal gland. Unilateral hyperplasia is referred to as primary adrenal hyperplasia (PAH). Familial hyperaldosteronism (FH) is also rare and two types have been described: FH type I and FH type II. FH type I, or glucocorticoid-remediable aldosteronism (GRA), is autosomal dominant in inheritance and associated with variable degrees of hyperaldosteronism, high levels of hybrid steroids (eg, 18-hydroxycortisol and 18-oxocortisol), and suppression with exogenous glucocorticoids. FH type II refers to the familial occurrence of APA or IHA or both.
Low Renin and High Aldosterone Primary Aldosteronism Aldosterone-producing adenoma (APA) ≈35% of cases Bilateral idiopathic hyperplasia (IHA) ≈60% of cases Unilateral (primary) adrenal hyperplasia ≈2% of cases Aldosterone-producing adrenocortical carcinoma < 1% of cases Familial hyperaldosteronism (FH) Glucocorticoid-remediable aldosteronism (FH type I) < 1% of cases FH type II (APA or IHA) < 2% of cases Ectopic aldosterone-producing adenoma or carcinoma < 0.1% of cases |
Low Renin and Low Aldosterone Hyperdeoxycorticosteronism Congenital adrenal hyperplasia 11β-Hydroxylase deficiency 17α-Hydroxylase deficiency Deoxycorticosterone-producing tumor Primary cortisol resistance |
Apparent Mineralocorticoid Excess (AME)/11β-Hydroxysteroid Dehydrogenase Deficiency Genetic Type 1 AME Type 2 AME Acquired Licorice or carbenoxolone ingestion (type 1 AME) Cushing syndrome (type 2 AME) |
Cushing Syndrome Exogenous glucocorticoid administration–most common cause Endogenous ACTH-dependent ≈85% of cases Pituitary Ectopic ACTH-independent ≈15% of cases Unilateral adrenal disease Bilateral adrenal disease Massive macronodular hyperplasia (rare) Primary pigmented nodular adrenal disease (rare) |
In the past, clinicians would not consider the diagnosis of primary aldosteronism unless the patient presented with spontaneous hypokalemia, and then the diagnostic evaluation would require discontinuing antihypertensive medications for at least 2 weeks. The spontaneous hypokalemia/no antihypertensive drug diagnostic approach resulted in predicted prevalence rates of less than 0.5% of hypertensive patients. However, it is now recognized that most patients with primary aldosteronism are not hypokalemic and that case-detection testing can be completed with a simple blood test (plasma aldosterone concentration [PAC]-to-plasma renin activity [PRA] ratio) while the patient is taking most antihypertensive drugs. Using the PAC-PRA ratio as a case-detection test, followed by aldosterone suppression confirmatory testing, has resulted in much higher prevalence estimates (5%-10% of all patients with hypertension) for primary aldosteronism.
The diagnosis of primary aldosteronism is usually made in patients who are in the third to sixth decade of life. Few symptoms are specific to the syndrome. Patients with marked hypokalemia may have muscle weakness and cramping, headaches, palpitations, polydipsia, polyuria, nocturia, or a combination of these. Periodic paralysis is a very rare presentation in Caucasians, but it is not an infrequent presentation in patients of Asian descent. The polyuria and nocturia are a result of a hypokalemia-induced renal concentrating defect and the presentation is frequently mistaken for prostatism in men. There are no specific physical findings. Edema is not a common finding because of mineralocorticoid escape. The degree of hypertension is usually moderate to severe and may be resistant to usual pharmacologic treatments. Although not common, primary aldosteronism may present with hypertensive urgencies. Patients with APA tend to have higher blood pressures than those with IHA. Hypokalemia is frequently absent; thus, all patients with hypertension are candidates for this disorder. In other patients, the hypokalemia only becomes evident with addition of a potassium-wasting diuretic (eg, hydrochlorothiazide, furosemide). Aldosterone excess also leads to a mild metabolic alkalosis because of increased urinary hydrogen excretion mediated both by hypokalemia and by the direct stimulatory effect of aldosterone on distal renal tubule acidification. Because of a reset osmostat, the serum sodium concentration tends to be high-normal or slightly above the upper limit of normal—this clinical finding is very useful when initially assessing the potential for primary aldosteronism.
Several studies have shown that patients with primary aldosteronism may be at higher risk than other patients with hypertension for target organ damage of the heart and kidney. When matched for age, blood pressure, and duration of hypertension, patients with primary aldosteronism have greater left ventricular mass by echocardiographic measurements than patients with other types of hypertension (eg, pheochromocytoma, Cushing syndrome, or essential hypertension). In patients with APA, the left ventricular wall thickness and mass decreases markedly 1 year after adrenalectomy. Patients presenting with either APA or IHA have a significantly higher rate of cardiovascular events (eg, stroke, atrial fibrillation, and myocardial infarction) than matched patients with essential hypertension who have similar degrees of hypertension duration and control.