Abbreviations
- ACTH Adrenocorticotropic hormone
- ADH Antidiuretic hormone (vasopressin)
- APACHE Acute physiology and chronic health evaluation
- CT Computed tomography
- DDAVP 1-deamino-8-D arginine vasopressin
- DKA Diabetic ketoacidosis
- FT4 Free thyroxine
- GFR Glomerular filtration rate
- IL Interleukin
- MRI Magnetic resonance imaging
- PTH Parathyroid hormone
- PTHrP Parathyroid hormone–related protein
- SIADH Syndrome of inappropriate antidiuretic hormone secretion
- T3 Triiodothyronine
- TPP Thyrotoxic periodic paralysis
- TSH Thyroid-stimulating hormone
Endocrine Emergencies: Introduction
Acute or chronic failure of an endocrine gland can occasionally result in catastrophic illness and even death. Thus, it is important to recognize and appropriately manage these endocrine emergencies. This chapter will discuss crises involving the thyroid, anterior pituitary, or adrenal glands; diabetes mellitus; and abnormalities in calcium, sodium, and water balance. Except where indicated, management recommendations are provided for adult patients. Studies in the general area of endocrine emergencies have been limited in size and number. In many instances, recommendations offered in this chapter are based on published expert opinion rather than scientific evidence.
Myxedema Coma
Myxedema coma is the end stage of untreated or inadequately treated hypothyroidism. The clinical picture is often that of an elderly obese female who has become increasingly withdrawn, lethargic, sleepy, and confused. The presentation is one of severe hypothyroidism, with or without coma (the term myxedema coma may, therefore, be a misnomer). The history from the patient may be inadequate, but the family may report that the patient has had thyroid surgery or radioiodine treatment in the past or that the patient has previously been receiving thyroid hormone therapy. Myxedema coma is most frequently associated with discontinuation of thyroid hormone therapy and less frequently as the first manifestation of hypothyroidism. Myxedema coma may be precipitated by an illness such as a cerebrovascular accident, myocardial infarction, or an infection such as a urinary tract infection or pneumonia. Other precipitating factors include gastrointestinal hemorrhage; acute trauma; excessive hydration; or administration of a sedative, narcotic, or potent diuretic drug.
The physical findings are not specific. The patient may be semicomatose or comatose with dry, coarse skin, hoarse voice, thin scalp and eyebrow hair, possibly a scar on the neck, and slow reflex relaxation time. There is marked hypothermia, with body temperature sometimes falling to as low as 24°C (75°F), particularly in the winter months. It is important to be alert to the presence of complicating factors such as pneumonia, urinary tract infection, ileus, anemia, hypoglycemia, or seizures. Fever may be masked by coexistent hypothermia. Often there are pericardial, pleural, or peritoneal effusions. The key laboratory tests are a low free thyroxine (FT4) and elevated thyroid-stimulating hormone (TSH). Note that in an emergency situation, serum TSH can be done in 1 hour. The TSH elevation may be less than predicted due to the presence of euthyroid sick syndrome or glucocorticoid or dopamine therapy. If the FT4 is low and the TSH is low-normal, consider central or pituitary hypothyroidism. Pituitary insufficiency can be confirmed with a low serum cortisol, impaired response to the cosyntropin stimulation test, and/or low follicle-stimulating hormone and luteinizing hormone. It is essential to check blood gases, electrolytes, creatinine, and an electrocardiogram in evaluating pulmonary, renal, cardiac, and central nervous system status. The combination of hypothermia, hyponatremia, and hypercapnia should increase suspicion of myxedema coma. It may be necessary to differentiate myxedema coma from the euthyroid sick syndrome associated with coma due to other causes. These patients may present with a low triiodothyronine (T3), normal or low TSH, but the FT4 (by dialysis) is normal.
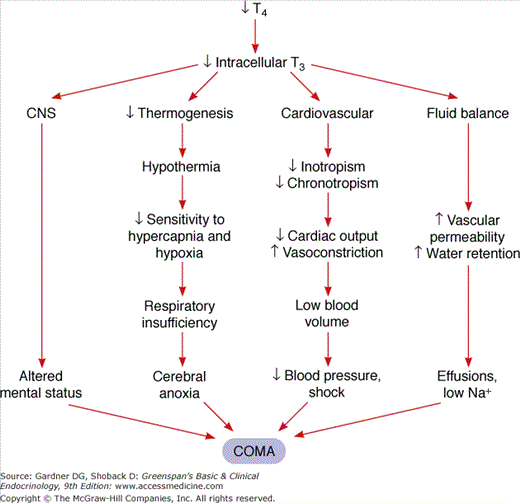
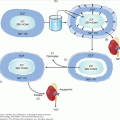
Myxedema coma is a complex problem involving a number of organ systems. The pathogenesis is presented in Figure 24–1. The decrease in serum T4 results in a lowering of intracellular T3. This can directly affect central nervous system function with altered mental status. The decrease in intracellular T3 causes decreased thermogenesis, resulting in hypothermia, which in turn causes decreased central nervous system sensitivity to hypercapnia and hypoxia. The resulting respiratory insufficiency induces cerebral anoxia and coma. At the same time, the decreased intracellular T3 results in decreased cardiac inotropism and chronotropism, decreased sensitivity to adrenergic stimuli, decreased cardiac output, and generalized vasoconstriction. This leads to a low-output state which, if untreated, culminates in decreased blood pressure and eventually shock and death. Finally, there is a change in fluid balance with increased water retention due to impaired renal perfusion as well as increased vascular permeability. These changes result in effusions and hyponatremia, which in turn contribute to the coma.
Management of myxedema coma involves much more than simply replacing T4 (Table 24–1). The patient is severely ill and usually is admitted to the intensive care unit (ICU) for intubation and ventilatory support. Oral medications may be poorly absorbed (due to gastric atony or ileus), and medications should be given intravenously if possible. A loading dose of 300 to 400 μg of T4 intravenously is given initially to saturate T4 binding sites in plasma-binding proteins. The patient is then maintained on 50 to 100 μg of T4 intravenously daily. In addition, small doses of T3 (eg, 10 μg intravenously every 6-8 hours) may be given over the first 48 hours, but this is usually not necessary, and it may contribute to untoward cardiovascular events. Its use should be restricted to younger patients without history of ischemic heart disease or cardiac arrhythmias. Water restriction is necessary to correct the hyponatremia, and intravenous glucose counteracts the tendency to hypoglycemia. It is essential to treat the precipitating illness (eg, pneumonia or urinary tract infection). The use of hydrocortisone is prudent because patients may have hypopituitarism or autoimmune polyglandular failure. Glucocorticoids are generally given in high doses until baseline cortisol or the results of rapid adrenocorticotropic hormone (ACTH) testing are available. If the initial serum cortisol is more than 30 μg/dL, steroid support is probably unnecessary. However, if serum cortisol is less than 30 μg/dL, hydrocortisone should be given intravenously in a dosage of 50 to 100 mg every 6 hours for the first 48 hours and the dose then tapered over the next 5 to 7 days while the pituitary-adrenal axis undergoes formal testing. Once myxedema coma is suspected in a hypothermic patient, external rewarming should be avoided, because this may cause redistribution of blood flow to subcutaneous tissues and cardiovascular collapse.
| (1) Admit to ICU for ventilatory support and for intravenous medications. |
| (2) Parenteral thyroxine: Give a loading dose of 300-400 μg IV, then 50-100 μg IV daily. (May also give liothyronine sodium, 10 μg IV every 8 h for the first 48 h if necessary.) |
| (3) Electrolytes: Water restriction for hyponatremia. Avoid fluid overload. |
| (4) Limit sedation. Appropriate reduction in drug dosage. |
| (5) Glucocorticoids: Controversial but necessary in hypopituitarism or polyglandular failure. Dosage: Hydrocortisone sodium phosphate or sodium succinate, 50-100 mg every 6 h initially and tapered downward over 1 wk. (If initial serum cortisol was >30 μg/dL, corticosteroids are unnecessary.) |
| (6) Hypothermia: Do not externally rewarm. |
Prior to the recognition of the need for intravenous T4 and for respiratory support, the mortality from myxedema coma was about 80%. Currently, the mortality is about 20% and is mostly due to the underlying or precipitating illness. Predictors of acute mortality include level of consciousness, lower Glasgow scores, and higher (acute physiology and chronic health evaluation) APACHE II scores, all indicative of the severity of the illness at presentation. Higher mortality is also associated with increased age, cardiac complications, and high-dose thyroid hormone replacement (≥500 μg/d of L-T4 or−≥75 μg/d of L-T3). The latter presumably reflects the increased metabolic demand attendant to high dose replacement in the setting of limited physiologic reserve. Persistent hypothermia and bradycardia, despite therapy, are associated with a poor prognosis.
Thyroid Storm
Thyroid storm, or thyrotoxic crisis, is an acute life-threatening exacerbation of thyrotoxicosis. It accounts for 1% to 2% of hospital admissions for thyrotoxicosis. It may occur in a patient with a history of Graves disease who has discontinued antithyroid medication or in a patient with previously undiagnosed hyperthyroidism. The clinical picture is that of an acute onset of hyperpyrexia (with temperature >40°C [104°F]), sweating, marked tachycardia often with atrial fibrillation, nausea, vomiting, diarrhea, agitation, tremulousness, and delirium. The presence of jaundice is considered a poor prognostic sign. Occasionally, the presentation is apathetic without the restlessness and agitation, but with symptoms of weakness, confusion, cardiovascular and gastrointestinal dysfunction, and hyperpyrexia. Some of the factors that may precipitate thyroid storm are listed in Table 24–2.
| Withdrawal of antithyroid drugs |
| Severe infection |
| Diabetic ketoacidosis |
| Myocardial infarction |
| Cerebrovascular accident |
| Cardiac failure |
| Surgery |
| Parturition |
| Trauma (eg, hip fracture) |
| Radioiodine (rare) |
| Drug reaction |
| Iodinated contrast medium |
The diagnosis is largely based in the clinical findings. Serum T4, free T4, T3, and free T3 are all elevated, and TSH is suppressed. These findings are not different from what is seen in other patients with hyperthyroidism, but the difference is in the setting. It is thought that thyroid storm represents an exacerbation of thyrotoxicosis associated with a shift of T4 from the bound to the free compartment with an increase in free T3 and T4, as well as an exaggerated response to a surge of catecholamines that results from the stress of the precipitating event. The cause of death is usually cardiac arrhythmia and failure. Liver function abnormalities are often seen, as is leukocytosis, even in the absence of infection.
The management of thyroid storm is summarized in Table 24–3. Patients are typically admitted to an ICU for cardiac monitoring. Initially it is important to block further synthesis and secretion of thyroid hormone, first with antithyroid drugs and then with iodide. One may give propylthiouracil, 150 mg every 6 hours orally or rectally, or methimazole, 20 mg every 8 hours orally or rectally. A few hours after initiation of antithyroid drug therapy, iodides may be started. Traditionally, saturated solution of potassium iodide, five drops twice daily, has been used, but currently iopanoic acid in a dose of 0.5 g twice daily orally or intravenously, or iohexol 0.6 g (2 mL of Omnipaque 300) intravenously twice daily is the treatment of choice. These drugs not only inhibit thyroid hormone synthesis but also block the conversion of T4 to T3, lowering the thyroid hormone level in the blood. Additional specific therapy includes β-adrenergic blockade with propranolol, 40 to 80 mg orally every 6 hours or 0.5 to 1 mg intravenously over 10 minutes every 3 hours, with continuous cardiac monitoring. In patients with asthma, the β1-selective antagonist esmolol can be given as a bolus dose (0.25-0.5 mg/kg over 10 minutes) followed by continuous intravenous infusion (0.05-0.1 mg/kg/min). The half-life of glucocorticoids is markedly reduced in severe thyrotoxicosis, so that adrenal support may be very useful. Dexamethasone also reduces conversion of T4 to T3 (as do the β blockers, especially propranolol). For this purpose, dexamethasone is given in a dosage of 2 mg every 6 hours for 48 hours, followed by tapering of the dose. Finally, cholestyramine or colestipol binds T4 in the gut interfering with its enterohepatic circulation and may help bring the circulating level of T4 down more quickly. Supportive therapy includes adequate fluids, oxygen, management of atrial fibrillation or heart failure, parenteral water-soluble vitamins, and a cooling blanket and acetaminophen for hyperpyrexia. Aspirin should be avoided, because it will displace T4 from thyroid hormone—binding globulin, resulting in an increase in FT4. Plasmapheresis or dialysis to remove FT4 has been reported to be useful in nonresponders, but this is rarely necessary.
| Supportive care |
Fluids Oxygen Cooling blanket Acetaminophen Multivitamins If indicated, antibiotics, digoxin |
| Specific measures |
Propranolol, 40-80 mg orally every 6 h Propylthiouracil, 150 mg every 6 h, or methimazole, 20 mg every 8 h; may be administered per rectum if oral route is unavailable Saturated solution of potassium iodide, 5 drops (250 mg) orally twice daily; or iopanoic acid, 0.5 g IV or orally twice daily; or iohexol, 0.6 g (2 mL of Omnipaque 300) IV twice daily Dexamethasone, 2 mg every 6 h Cholestyramine or colestipol, 20-30 g/d |
Therapy for thyroid storm has improved markedly, so the mortality has dropped from 100% in the 1920s to about 20% to 30% in recent series. However, because storm is often associated with other underlying medical problems, it still represents a serious medical complication.
Thyrotoxic Periodic Paralysis
Thyrotoxic periodic paralysis (TPP) is a rare but frightening thyroid emergency. The usual clinical presentation is of an Asian male (male:female ratio approximately 17:1) with symptoms of untreated hyperthyroidism who awakens at night or in the morning with flaccid ascending paralysis. Typically, there is a history of vigorous exercise and/or a large high-carbohydrate meal before retiring. There is usually no family history of periodic paralysis, but there may be a family history of autoimmune thyroid disease. The paralysis initially involves the lower extremities but progresses to the girdle muscles, followed by the upper extremities. Proximal muscle groups are affected to a greater extent than distal. Facial and respiratory muscles are usually spared. Sensory function, bowel and bladder function are not affected. Deep tendon reflexes are depressed or absent. The acute episode may be complicated by cardiac arrhythmias due to the concomitant presence of hypokalemia. The illness has also been reported to occur in Native Americans, African Americans, and in individuals of Mexican or South American descent, but these ethnic groups are affected rarely.
The differential diagnosis of TPP includes familial periodic paralysis, Guillain-Barré syndrome, and acute intermittent porphyria. The diagnosis is based on the absence of a family history, the characteristic presentation, the presence of hyperthyroidism due either to Graves disease or toxic nodular goiter (other types of hyperthyroidism have been implicated as well), and usually a low serum potassium level.
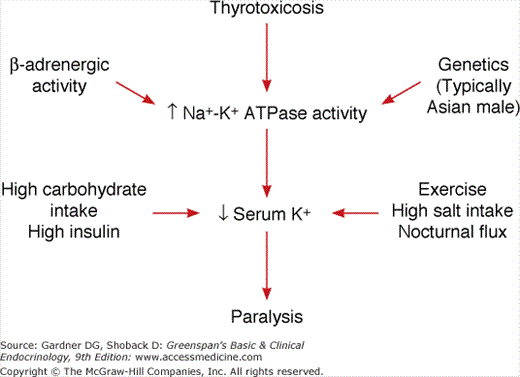
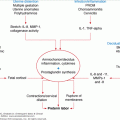
The pathogenesis is summarized in Figure 24–2. Thyrotoxicosis, increased β-adrenergic activity, and an assumed genetic predisposition, perhaps involving the potassium channel Kir2.6, which together with increased Na+-K+ ATPase activity leads to increased intracellular potassium concentrations. A high-carbohydrate meal with increased insulin secretion and glycogen deposition, vigorous exercise high salt intake, and the normal nocturnal potassium flux serve to drive serum potassium levels even lower, resulting in flaccid neuromuscular paralysis. Note that there is no loss of total body potassium, merely a shift from the extracellular to the intracellular space. Consequently, aggressive potassium repletion, particularly through the parenteral route, is discouraged because this frequently results in significant hyperkalemia as potassium redistributes across cellular compartments following resolution of the attack. Hypophosphatemia and hypomagnesemia may be present. This is also assumed to reflect intracellular sequestration. The electromyogram, performed while the patient is experiencing weakness, shows myopathic changes with reduced amplitude of compound muscle action potentials. These do not change in amplitude after administration of intra-arterial low-dose epinephrine (distinguishes from familial periodic paralysis). Electrocardiograms show changes associated with hypokalemia, tachycardia, increased QRS voltage, first-degree heart block, and, on occasion, serious ventricular arrhythmias.
The management of this problem is presented in Table 24–4. Propranolol in doses of 60 mg every 6 hours blocks the β-adrenergic stimulation of Na+-K+ ATPase. Antithyroid drug therapy should be started immediately, even though it takes time to bring the patient into a euthyroid state. Oral potassium, if indicated, should be administered cautiously. It is particularly important to be cautious if administering intravenous potassium, which may raise total body potassium to toxic levels as the episode resolves. One should also avoid intravenous glucose, which stimulates insulin secretion and worsens hypokalemia, and β-adrenergic agonists such as isoproterenol, which promote movement of potassium into the intracellular compartment and exacerbate the problem. There is no role for potassium supplementation in preventing attacks. Acetazolamide, which has been shown to reduce frequency of attacks in familial periodic paralysis, may worsen attacks of thyrotoxic periodic paralysis and should be avoided. With appropriate treatment, recovery is rapid, and once the thyrotoxicosis is controlled, the paralysis will not recur.
(1) Oral potassium supplement (if needed); monitor serum K+ (2) Oral propranolol (60 mg every 6 h) (3) Antithyroid drug therapy |
| Avoid: |
IV potassium IV glucose (ie, crystalloid fluid replacement only) β-Adrenergic agonists (eg, isoproterenol) |
Amiodarone-Induced Thyrotoxicosis
Amiodarone is a benzofuran derivative that is widely used in the treatment of cardiac arrhythmias. It contains two atoms of iodine per molecule, which represents 37.5% iodine by weight. The compound is stored in adipose tissue and has a half-life in the body of 2 to 3 months, with gradual and continuous release of iodide. The usual daily maintenance dose of amiodarone of 200 to 400 mg/d releases 6,000 to 12,000 μg of iodine daily, which when compared to the normal daily requirement of about 150 μg of iodide represents an enormous iodide load.
The structure of amiodarone resembles that of T3, and it is thought that part of the cardiac depressant effect of amiodarone may be due to binding to and blocking the T3 receptor in cardiac muscle. However, the effect of amiodarone on the thyroid gland is different and is due in part to a direct effect of iodine on the thyroid cell—to inhibit or stimulate hormone synthesis—and a cytotoxic effect of amiodarone on the follicular cell—inducing destruction of the cell and release of stored hormone. Thus, the drug may induce hypothyroidism, which is easily managed by thyroxine replacement, or hyperthyroidism, which, because of the underlying heart disease, is much more difficult to manage and may represent a true thyroid emergency. Two mechanisms have been suggested to explain the development of hyperthyroidism: (1) the high iodine level in a multinodular gland, or in the gland of a patient with latent Graves disease, or even in a previously normal gland, can induce hyperthyroidism (see Chapter 7), and (2) the toxic effect of amiodarone itself may cause acute and chronic thyroiditis with release of T4 and T3 into the circulation and severe thyrotoxicosis. Risk factors for amiodarone-induced thyrotoxicosis include the presence of autoimmune thyroiditis and baseline elevation of TSH levels.
The patient with amiodarone-induced thyrotoxicosis may have been on the drug for months. Thyrotoxicosis typically appears 6 to 12 months after the onset of treatment. The underlying heart disease gradually worsens with increasingly frequent episodes of arrhythmia and heart failure. At the same time there may be weight loss, heat intolerance, increased nervousness, and marked muscle weakness. On physical examination, one may find nontender nodular or diffuse thyroid enlargement, tachycardia with or without atrial fibrillation, tremor, hyperreflexia, and, occasionally, lid lag and stare. Laboratory findings are unique because amiodarone inhibits the conversion of T4 to T3. Thus, even in the euthyroid patient taking amiodarone, total T4 may be elevated while FT4 and TSH are normal. In the hyperthyroid patient, the FT4 is markedly elevated and TSH is less than 0.01 mU/L (below the detection limit of the assay). Radioiodine uptake in the iodide-loaded patient is low. It has been difficult to distinguish thyrotoxicosis due to follicular cell hyperfunction from that due to follicular cell destruction. Thyroid ultrasound with color Doppler studies may show increased circulation with hyperfunction and decreased blood flow with thyroiditis. Also, the cytokine interleukin (IL)-6 is low in patients with hyperfunction, whereas it is markedly elevated in patients with thyroiditis. However, these tests have not proven sufficiently robust to separate these two disorders.
Management of amiodarone-induced hyperthyroidism is difficult (Table 24–5). Ideally, amiodarone should be discontinued, but often it cannot be stopped because of the underlying heart disease, and even if it is discontinued, the iodine load persists for several months. Further synthesis of T4 should be blocked with methimazole in a dosage of 40 to 80 mg/d or propylthiouracil in doses of 400 to 800 mg/d. β-Adrenergic blockade, if needed (amiodarone, itself, has some β-blocking activity), should be instituted with propranolol or a comparable drug if cardiac status permits it. Potassium perchlorate in a dosage of 250 mg every 6 hours blocks further iodine uptake and lowers intrathyroidal iodide content. Aplastic anemia has occurred in patients on high-dose or long-term potassium perchlorate therapy, so that use of this medication has usually been limited to 1 month. Iopanoic acid (0.5 g orally twice daily) can be used to decrease T4 to T3 conversion. Cholestyramine or colestipol in a dosage of 20 to 30 g/d binds T4 and T3 in the gut and brings blood levels down more quickly. If there is reason to suspect thyroiditis (elevated serum IL-6 or decreased blood flow on ultrasound), corticosteroid therapy often yields dramatic results. Prednisone is given in a dosage of 40 mg/d for 1 month, gradually tapering the dose over the following 2 months. In those cases where a mixed etiology is suspected (ie, hyperfunction plus thyroiditis), a combination of steroids, antithyroid drugs, and β blockers can be used. If medical therapy is unable to control the disease, thyroidectomy results in a permanent cure and may be used as a last resort. Iopanoic acid (see above) can be used to prepare the patient for surgery. Perioperative mortality may be as high as 8% to 9% due to the underlying cardiac disease.
| (1) Stop amiodarone if possible |
| (2) Institute β-blocker therapy if possible |
| (3) Antithyroid drugs: Methimazole, 40-60 mg/d |
| (4) Potassium perchlorate, 200 mg every 6 h |
| (5) Cholestyramine or colestipol, 20-30 g/d |
| (6) Prednisone, 40 mg/d, for acute thyroiditis (consider monitoring IL-6 levels) |
| (7) Thyroidectomy |
Acute Adrenal Insufficiency
Acute adrenal insufficiency usually occurs as an acute illness in a patient with chronic adrenal insufficiency (see Chapter 9). The chronic adrenal insufficiency may be primary, due to destruction of the adrenal glands associated with autoimmune adrenalitis, adrenal leukodystrophy or, rarely, tuberculosis, fungus, or metastatic malignancy. Chronic adrenal insufficiency may also be secondary to pituitary or hypothalamic disease. Acute adrenal insufficiency may also occur with bilateral adrenal hemorrhage in a previously healthy individual during the course of septicemia with disseminated intravascular coagulopathy or in a patient receiving anticoagulant therapy. In the patient with known adrenal insufficiency, an acute crisis may be precipitated by inadvertent omission of steroid medication or by the concurrent development of a precipitating illness such as severe infection, acute myocardial infarction, cerebrovascular hemorrhage or infarction, surgery without adrenal support, or severe acute trauma. Acute adrenal insufficiency may also be precipitated by the sudden withdrawal of steroids in a patient previously on long-term steroid therapy with associated adrenal atrophy (ie, secondary adrenal insufficiency). Finally, administration of drugs impairing adrenal hormone synthesis such as ketoconazole, aminoglutethimide, etomidate, or mitotane—or drugs increasing steroid metabolism such as phenytoin or rifampin—may precipitate an adrenal crisis.
The patient presents with an acute onset of nausea, vomiting, hyperpyrexia, abdominal pain, dehydration, hypotension, and shock. A clue to the diagnosis of primary adrenal insufficiency is the presence of pigmentation in unexposed areas of the skin, particularly in the creases of the palms and in the buccal mucosa. The differential diagnosis includes consideration of other causes of cardiovascular collapse, sepsis, and intra-abdominal abscess. Failure of the hypotension to respond to pressors is suggestive of adrenal insufficiency and is an indication for a trial of glucocorticoid therapy.
Primary adrenal insufficiency is characterized by hyponatremia and hyperkalemia. However, in situations of adrenal crisis, the hyponatremia may be obscured by dehydration. Random serum cortisol determinations are not helpful unless the levels are very low (<5 μg/dL [138 nmol/L]) during a period of great stress. The key diagnostic test is failure of serum cortisol to rise above 20 μg/dL (552 nmol/L) 30 minutes after intravenous injection of 250 μg synthetic ACTH (cosyntropin) (see Chapter 9 for details). This test functions best in the diagnosis of primary versus secondary adrenal insufficiency. At a specificity of 95%, sensitivities are 97% and 57%, respectively (see Chapter 3). Interpretation of the test, particularly in the evaluation of secondary adrenal insufficiency, is complicated in the presence of hypoalbuminemia, a marker for reduced protein-bound cortisol in plasma. In this setting, serum-free cortisol levels provide a more accurate assessment of adrenal function. Random serum-free cortisol of >1.8 μg/dL and a Cortrosyn-stimulated value of >3.1 μg/dL in critically ill patients is considered normal. There has been increased interest in using a more physiologic dose of ACTH to perform this stimulation test (1 μg vs 250 μg); however, a recent comparison suggests no particular advantage in the performance characteristics of the low-dose test. Basal serum ACTH is elevated (>52 pg/mL [>11 pmol/L]) in patients with primary adrenal insufficiency but not in patients with secondary adrenal insufficiency due to pituitary or hypothalamic disease. Computed tomography (CT) or sonography of the abdomen reveals adrenal enlargement in patients with adrenal hemorrhage, active tuberculosis, or metastatic malignancy. Atrophy of the adrenals is associated with chronic adrenal insufficiency.
The management of adrenal crisis is outlined in Table 24–6. Hydrocortisone should be administered in a dosage of 100 mg intravenously followed by 50 to 75 mg every 6 hours thereafter. Fluids and Na+ should be replaced with several liters of 5% glucose in normal saline. After the first 24 hours, the dose of intravenous hydrocortisone can be slowly reduced, but intravenous doses should be given at least every 6 hours because of the short half-life (1 hour) of hydrocortisone in the circulation. When the patient can tolerate oral feedings, hydrocortisone can be given orally, but the first oral dose should overlap the last intravenous dose. Alternatively, hydrocortisone can be administered as a continuous infusion at the rate of 10 mg/h for the first 24 hours, followed by a gradual decrease in the dose. Mineralocorticoid is not necessary during the acute replacement period since enough NaCl and glucocorticoid are being administered to treat the mineralocorticoid deficiency. However, in patients with chronic primary adrenal insufficiency, mineralocorticoid supplementation is necessary when shifting to an oral maintenance program (see Chapter 9). After steroid therapy has been instituted, it is extremely important to evaluate and treat the illness that may have precipitated the acute crisis (eg, infection, myocardial infarction).
| (1) Hydrocortisone sodium phosphate or sodium succinate, 100 mg IV stat and then 50-75 mg IV every 6 h for 24 h. Then taper slowly downward over the next 72 h, giving the drug every 4-6 h IV. When patient is tolerating oral feedings, shift to oral replacement therapy, overlapping the first oral and last intravenous doses. |
| (2) Replace salt and fluid losses with several liters of 5% glucose in normal saline IV. |
| (3) Patients with primary adrenal insufficiency may require mineralocorticoid (fludrocortisone) when shifted to oral hydrocortisone maintenance therapy. |
| (4) Diagnose and treat the illness that precipitated the acute crisis. |
Prevention of acute adrenal insufficiency in patients with chronic adrenal insufficiency exposed to mild stress can be addressed merely by doubling the daily steroid dose until the condition resolves followed by rapid titration back to prestress doses. More severe stress (eg, severe infection) requires intravenous hydrocortisone in dosages as outlined above or administration of dexamethasone sodium phosphate, 4 mg intramuscularly every 24 hours for two doses. Dexamethasone would replace glucocorticoids but not mineralocorticoid and would not be adequate in the presence of severe dehydration.
Studies of patients with septic shock have identified a subgroup that may have relative adrenal insufficiency (ie, decreased adrenal reserve). These patients were identified based on a limited increment (ie, the difference between basal and post-ACTH cortisol levels) rather than the absolute level of post-ACTH cortisol. A recent randomized trial (CORTICUS [Corticosteroid Therapy of Septic Shock] study) demonstrated that hydrocortisone therapy did not improve survival or reversal of shock in patients with septic shock, regardless of their response in the Cortrosyn stimulation test. However, hydrocortisone did hasten reversal of shock in those patients in whom shock was reversed. At the present time, the utility of steroid therapy in the management of vasopressor-dependent septic shock remains controversial.
Pituitary Apoplexy
Pituitary apoplexy is a rare but frightening syndrome of violent headache, visual and cranial nerve disturbances, and mental confusion, resulting from hemorrhage or infarction of a pituitary tumor or, more rarely, a normal pituitary gland.
Pituitary apoplexy usually occurs as a sudden crisis in a patient with a known or, more rarely, previously unrecognized pituitary tumor. However, it may occur in a normal gland during or after parturition. Risk factors include cardiac surgery, dynamic pituitary function testing, head trauma, or anticoagulation therapy. The patient presents with severe headache and visual disturbances, often a bitemporal hemianopia due to compression of the optic chiasm. There may be oculomotor defects, either bilateral or unilateral, due to extension of the hemorrhage into the cavernous sinus(es) where cranial nerves II, III, IV, and VI are located. Often there are meningeal symptoms with stiff neck and mental confusion, so that the differential diagnosis includes subarachnoid hemorrhage and meningitis. Finally, there may be symptoms of acute secondary adrenal insufficiency with nausea, vomiting, hypotension, and collapse. A more subacute/chronic presentation (ie, subclinical hemorrhage followed by partial resolution) may be dominated by symptoms of hypopituitarism.
The diagnosis of pituitary apoplexy is best approached with magnetic resonance imaging (MRI) of the cranium with pituitary views. Pituitary enlargement and signs of hemorrhage are diagnostic. In the acute setting, hormonal studies are of academic interest only since therapy should include glucocorticoid support regardless of the acute findings. After appropriate acute therapy, evaluation of anterior and posterior pituitary function is indicated to evaluate the possibility of permanent hypopituitarism.
Management involves both hormonal and neurosurgical therapy. High-dose dexamethasone, 4 mg twice daily, provides both glucocorticoid support and relief of cerebral edema. Alternatively, hydrocortisone 50 mg IV every 6 hours can be used. Trans-sphenoidal pituitary decompression often provides dramatic relief of visual and extraocular motor dysfunction and in the level of consciousness. Pituitary hormone secretion also may resolve particularly in those with normal or elevated prolactin levels. The need for emergent surgery is controversial and should be approached on a case-by-case basis. Conservative medical management in less severely affected patients has been shown to result in recovery of both neurological and endocrine function. After the acute episode has subsided, the patient must be evaluated for the possibility of multiple pituitary hormonal deficiencies (see Chapter 4).
Diabetic Ketoacidosis
Diabetic ketoacidosis (DKA) occurs in a setting of absolute or relative insulin deficiency. Estimated annual incidence is 4 to 8 episodes per 1000 patients with diabetes. Specific clinical settings should generate a high index of suspicion for the disorder. Interruptions of normal insulin delivery due to purposeful reduction in insulin dosage or interference with the delivery system (eg, kinking in pump tubing) are frequent precipitating events, as are reduced insulin sensitivity in the setting of systemic infection, myocardial infarction, burns, trauma, or pregnancy. In a significant percentage of patients, DKA is the presenting feature of diabetes. In these instances, clinical suspicion and accurate interpretation of the initial laboratory studies will usually lead to the correct diagnosis. Measurement of HbA1<!–sb>C (hemoglobin A<sb>1<!–sb>C) levels may help in assessing the chronicity of the diabetes. Mortality in DKA is less than 5% in experienced centers. Prognosis worsens at the extremes of age and in the presence of coma or hypotension.
DKA is characterized metabolically by two prominent features: hyperglycemia and ketoacidosis (see Chapter 17). Patients with DKA present with evidence of volume contraction (eg, dry mucous membranes, thirst, orthostatic hypotension) and labored breathing (Kussmaul respiration) related to the underlying acidosis. The breath often has a fruity odor, reflecting the presence of acetone. Patients may have abdominal pain mimicking an acute abdomen, nausea, and vomiting. The latter symptoms may be related to elevated gastrointestinal prostaglandins that accrue in the presence of insulin deficiency. Presentation may be dominated by symptoms of the precipitating illness (eg, urinary tract infection, pneumonia, or myocardial infarction).
Plasma glucose levels are elevated, usually to over 250 mg/dL. This reflects impairment in glucose utilization (see discussed earlier), increased gluconeogenesis and glycogenolysis, and reduced renal clearance of glucose in the setting of decreased glomerular filtration rate (GFR). Osmotic diuresis related to glucose excretion results in reduction in intravascular volume and depletion of total body water, sodium, potassium, phosphate, and magnesium. In general, the relative depletion of water is roughly twice that of the solutes it contains. Hypertonicity in the extracellular fluid compartment, although typically not as severe as that seen in hyperosmotic nonketotic coma (see later), can be significant. Calculated plasma osmolalities greater than 340 mOsm/kg are associated with coma. Plasma osmolality—rather than acidemia—correlates most closely with state of consciousness in DKA (Figure 24–3).
Figure 24–3
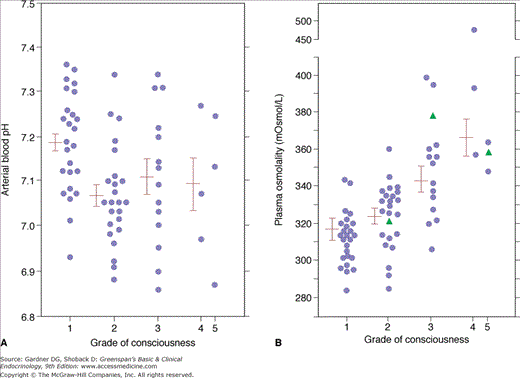
A: Relationship between state of consciousness and blood pH in patients with diabetic ketoacidosis. B: Relationship between state of consciousness and plasma osmolality in diabetic ketoacidosis. Note that the state of consciousness correlates with plasma osmolality rather than blood pH.
(Reproduced, with permission, from Fulop M, et al. Ketotic hyperosmolar coma. Lancet. 1973;2:635.)
Arterial blood pH is low and, in the absence of coexistent respiratory disease, is partially compensated by a reduction in Pco2. The acidosis is metabolic in origin and accompanied by an anion gap that is calculated by subtracting the combined concentrations of chloride and bicarbonate from serum sodium concentration. Anion gaps greater than 12 mEq/L are considered abnormal. Keto acids account for most of the unmeasured anions that generate the abnormal gap, although under conditions of extreme volume contraction and hypoperfusion, lactate accumulation may also contribute. Levels of serum and urinary ketones (measured using the nitroprusside reagent) are typically high in DKA. It should be recalled, however, that this reagent reacts strongly only with acetoacetate, less strongly with acetone (which is not a keto acid and does not contribute to the anion gap), and not at all with β-hydroxybutyrate. Thus, paradoxically, the most extreme levels of ketoacidosis may be accompanied by relatively modest levels of ketones measured by this method. As a corollary of this, resolution of severe DKA may be linked to transient increases in measurable ketone levels as β-hydroxybutyrate is converted to the more readily detectable acetoacetate.
Serum sodium levels may be high, normal, or low, but in all instances total body sodium is depressed. Estimates of depletion range from 7 to 10 mEq/kg body weight. As blood glucose levels rise in DKA, they create an osmotic gradient that draws water, as well as intracellular solutes, into the extracellular space. This results in moderate hyponatremia, which can be corrected to account for the dilutional effect of the transmembrane flux of water by adding 1.6 mEq/L (more recent studies suggest that the correction factor is closer to 2.4 mEq/L) to the sodium concentration for every 100 mg/dL increment in plasma glucose above a basal concentration of 100 mg/dL (see below). The decrease in serum sodium partially offsets the increase in tonicity that accompanies the elevation in plasma glucose. This results in a net increase in plasma osmolality of 2 mOsm/kg H2O per 100 mg/dL elevation in plasma glucose: 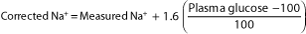
Total body potassium levels are also severely depleted in DKA to an average of 5 to 7 mEq/kg body weight. This results from a number of factors, including exchange of intracellular potassium for extracellular hydrogen ion, impaired movement of K+ into cells in the insulinopenic state, increased urinary potassium excretion secondary to the osmotic diuresis and, in those instances where intravascular volume contraction is present, secondary hyperaldosteronism. Serum potassium levels may be high, normal, or low depending on the severity and duration of DKA, the status of extracellular fluid volume, and the adequacy of renal perfusion and excretory function. A low serum potassium at presentation generally indicates severe potassium deficiency and, in the presence of adequate renal function, is an indication for early and aggressive repletion (see below). Potassium depletion can result in muscle weakness and cardiac arrhythmias, including ventricular fibrillation.
The H+
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


