The hereditary paraganglioma, MEN1, MEN2, and hereditary thyroid cancer syndromes are clinically discernable and genetically distinct. The first 3 syndromes have been well characterized in the past 10 to 15 years. Recognizing these 3 syndromes and using a multidisciplinary team approach creates valuable opportunities for early diagnosis, reduction of morbidity and mortality, and avoidance of surgical misadventures. Hereditary paraganglioma has parent-of-origin effects and gene-environment interactions that indicate its evolution, and the syndrome sheds light on the role of mitochondria and energy metabolism in cancer. This article delineates the clinical presentation and practical management issues and summarizes the history, gene discovery, and molecular insights for each syndrome.
The hereditary paraganglioma, MEN1, MEN2, and hereditary thyroid cancer syndromes overlap to some extent, but all are clinically discernable and genetically distinct. The first 3 syndromes have been well characterized in the past 10 to 15 years following identification of their molecular underpinnings, and genetic testing plays an indispensable role in clinical management in children and adults. The physician who recognizes these syndromes and uses a multidisciplinary team approach can take advantage of valuable opportunities for early and presymptomatic diagnosis, reduction of morbidity and mortality, and avoidance of surgical misadventures. Hereditary paraganglioma is a fascinating syndrome with parent-of-origin effects and gene-environment interactions that indicate a stamp placed on the human genome by natural selection, and the syndrome sheds light on the role of mitochondria and energy metabolism in cancer. MEN1 probably has the widest pleiotropy of any hereditary cancer syndrome. MEN2 is notable for remarkably precise genotype-phenotype correlations at the level of the codon that determine timing of surgical prevention. This article delineates the clinical presentation and practical management issues and summarizes the history, gene discovery, and molecular insights for each syndrome.
The clinical recognition and description of multiple endocrine neoplasia (MEN) and its subtypes began in the 1900s. The importance of managing patients and families in the context of multidisciplinary teams including genetics professionals cannot be overemphasized for MEN type 1, MEN type 2, and hereditary paraganglioma syndromes.
Hereditary paraganglioma syndrome
The 1933 report by Chase of carotid body tumors diagnosed in 2 sisters in their 20s, 1 of whom had bilateral disease, coupled with a report in the same year of paragangliomas in 3 sisters and several publications in the following decades, shed light on the hereditary nature of paragangliomas, at least in some cases. Autosomal dominant inheritance was suggested in 1949 and in subsequently reported pedigrees. A review of 88 familial and 835 nonfamilial carotid body tumor cases in the literature showed a high rate of bilateral disease in familial (31.8%) versus sporadic (4.4% cases), and an equal sex distribution in familial cases, consistent with autosomal dominant inheritance. A medical record review of 222 carotid body tumors at 12 medical centers in the United States suggested the existence of a multiple primary tumor syndrome in familial cases that manifested bilateral disease, other extra-adrenal paragangliomas, and earlier age of onset. Efforts then concentrated on the collection of families for linkage mapping, culminating in the discovery of the first hereditary paraganglioma gene, SDHD , and candidate gene analyses soon followed, leading more recently to predictive clinical genetic testing, a better understanding of genotype-phenotype relationships and gene-environment interactions, and important insights into the role of mitochondrial metabolism in cancer and the mechanism of parent-of-origin effects seen in the PGL1 and PGL2 syndromes.
Nonchromaffin paragangliomas are rare, occurring in about 1:30,000 to 1:100,000 individuals in the general population. Carotid body tumors are associated with conditions of low oxygen tension, such as emphysema, living at high altitude, and cyanotic congenital heart disease, and cluster in low-altitude countries.
Pathology, Anatomy, and Biochemistry
Pheochromocytomas and extra-adrenal paragangliomas are tumors derived from neural crest tissues or organs, termed paraganglia. Classification is based on anatomic considerations; lesions cannot be distinguished using histologic features. The term pheochromocytoma is reserved by the World Health Organization (WHO) for tumors that arise from chromaffin cells of the adrenal medulla, a prototypical sympathetic nervous system paraganglion. Extra-adrenal paragangliomas derive from either the sympathetic nervous system or the parasympathetic nervous system. The term extra-adrenal pheochromocytoma is imprecise.
The sympathetic nervous system includes the organ of Zuckerkandl (located at the origin of the inferior mesenteric artery and involved in fetal and newborn catecholamine metabolism) as well as chromaffin cells clustered in the paravertebral chain, aortic bifurcation, kidney, liver hila, bladder, and mediastinum. The parasympathetic nervous system is distributed from the middle ear and skull base to the pelvic floor, and includes the carotid body, a tiny organ located at the carotid bifurcation, which is the major oxygen sensor. Sympathetic extra-adrenal paragangliomas are often hormonally active. Parasympathetic extra-adrenal paragangliomas are usually located in the head and neck, and only about 1% are hormonally active.
Terminology for head and neck paragangliomas is varied and includes chemodectoma, carotid body, jugular, vagal, tympanic tumor, and glomus tumor. Pheochromocytomas (of the adrenal medulla) often have an adrenergic profile, whereas extra-adrenal paragangliomas (when active) typically have a noradrenergic profile. Pheochromocytomas are more typical of MEN2A, MEN2B, von Hippel-Lindau syndrome (VHL) (particularly type 2), and neurofibromatosis type 1 (NF1) than hereditary paraganglioma syndromes, but overlap exists. For example, head and neck paragangliomas are an occasional feature of MEN2, VHL, and NF1, and the hereditary paraganglioma syndromes can manifest sympathetic extra-adrenal paragangliomas and pheochromocytomas. Hereditary nonchromaffin paragangliomas are slow-growing, highly vascularized tumors, most of which are benign. The WHO defines malignancy for pheochromocytomas and extra-adrenal paragangliomas not by local invasion but as metastatic spread to distal sites (which are typically devoid of paraganglionic tissue; generally bone, liver, and lung) or clearly identifiable lymph nodes. Adequately resected MEN2 pheochromocytomas do not typically recur, whereas SDHB -related tumors have about a 50% rate of metastasis.
Inheritance and Genetics
Paraganglioma families typically have an autosomal dominant pattern with age-dependent penetrance. In certain linkage groups, individuals manifesting disease inherit the trait solely through the paternal lineage, suggestive of maternal imprinting. The PGL1 ( SDHD ) and PGL2 loci both show parent-of-origin effects consistent with maternal imprinting. Parent-of-origin effects are shown in Fig. 1 , and have important implications for screening. A single case of an affected offspring following maternal transmission of an SDHD mutation has been reported. The PGL3 ( SDHC ) and PGL4 ( SDHB ) loci follow classic autosomal dominant inheritance for tumor predisposition.
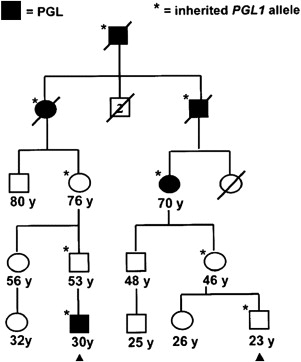
Early linkage studies mapped the PGL1 gene (later shown to be SDHD ) to 11q23-qter, PGL2 ( SDHAF2 / SDH5 ) to 11q13, and PGL3 ( SDHC ) to 1q21-q23. PGL4 ( SDHB ) maps to 1p36.1-p35. Baysal and colleagues identified the first paraganglioma gene in PGL1 kindreds as SDHD , which encodes the D subunit of heterotetrameric succinate dehydrogenase (SDH; mitochondrial complex II) enzyme complex, a component of the respiratory chain and the Krebs cycle. Following this discovery, the other SDH gene subunits became prime candidates for the other genetic loci. Mutations in SDHC occur in PGL3 families and mutations in SDHB in PGL4 families. However, the A subunit of SDH maps to chromosome 5p15, not to the remaining PGL2 locus at 11q13. SDHA mutations are one cause of Leigh syndrome, infantile subacute necrotizing encephalopathy. SDHAF2 / SDH5 assignment to the PGL2 locus was accomplished in yeast models and maps to 11q13, the PGL2 locus.
Clinical Presentation and Diagnosis
Paragangliomas of the head and neck typically present as slowly growing masses that can be asymptomatic for years or even decades. On careful questioning or evaluation, what may seem to be an isolated case can lead to unmasking of a family history of neck mass or other related growth. About 10% of patients present with cranial nerve palsy, most commonly involving the vagal nerve. Left untreated, about 75% of patients develop cranial nerve palsies, providing the potential for presymptomatic diagnosis, intervention, and prevention of serious morbidity. Symptoms include tinnitus, hearing loss, hoarseness, vocal cord paralysis, cough, aspiration, dysphagia, pharyngeal fullness, facial paralysis, problems moving the tongue, or pain. Vagal paragangliomas are located behind the ramus of the mandible and can project into the oropharynx. Magnetic resonance imaging (MRI) is diagnostic based on tumor vascularity and location, characterizes lesions for surgical planning, and assesses multifocality. Incisional biopsy of the highly vascular head and neck paragangliomas that are intimate with neurovascular structures is dangerous, usually unnecessary for diagnosis, and should be avoided; however, when a radiologic diagnosis is unclear, ultrasound-guided fine-needle aspiration can be used.
Pheochromocytomas and functional extra-adrenal paragangliomas may present with the classic triad of episodic headache, diaphoresis, and tachycardia, or with other symptoms. More typically, the classic triad is not seen. Persistent hypertension and normotension are also consistent with the diagnosis of a functional chromaffin cell tumor. Diagnosis of pheochromocytoma can be performed through biochemical testing and localization by imaging. Biochemical testing entails measurement of either 24-hour urine catecholamines and metanephrines or fractionated plasma-free metanephrines; the rationale for choosing one rather than the other has been discussed.
Genotype-Phenotype Correlations, Penetrance, and Expressivity
SDHD and SDHB mutations are more often associated with a positive family history than SDHC mutations. SDHC mutation carriers have a lower risk of pheochromocytoma, multifocality, and malignant transformation. The median age of onset for SDHC is slightly higher than for SDHD and SDHB and the earliest tumor reported in SDHC is at 13 years of age. Age at diagnosis of the first tumor often occurs by age 10 years for SDHB (earliest reported age, 7 years) and penetrance is 25% by age 20 years. Age less than 10 years for a first tumor is rarer, but not unknown for SDHD (earliest reported age, 5 years) and penetrance is 15% by age 20 years. The predominant phenotype for SDHB is extra-adrenal paraganglioma, and renal cell carcinoma is an important sign of SDHB . SDHB is associated with a 41% prevalence of malignant tumors, which may be related to a trend toward malignancy in extra-adrenal paragangliomas. The more severe phenotype of SDHB may be related to its role as part of the catalytic complex. Carriers with nonsense/splicing mutations developed symptoms 8.5 years earlier than missense mutation carriers, and a trend was seen toward pheochromocytoma development with nonsense mutations. Altitude correlates with tumor type. Mutations in the 5′ portion of the SDHD gene are correlated with pheochromocytoma, whereas 3′ mutations are associated with head and neck paragangliomas.
Several reports indicate that renal cell carcinoma is a component tumor for SDHB and renal oncocytoma has also been reported. Thyroid cancer, in particular the papillary type, might be part of the SDH gene spectrum, but this is not well established. In a large fraction of cases, the Carney-Stratakis syndrome (dyad) of paragangliomas and gastrointestinal stromal tumors (GIST) can be attributed to germline SDHD , SDHB , and SDHC mutations. SDHD and SDHB germline mutations or variants have been found in patients with a Cowden syndrome–like presentation negative for PTEN mutations. In these patients, the tumor spectrum seems to include breast, papillary thyroid, and renal cancers.
Diagnostic Criteria for Molecular Genetic Testing
There is an emerging consensus of opinion that all patients with a single head and neck paraganglioma, extra-adrenal paraganglioma, or nonsyndromic pheochromocytoma should undergo molecular genetic testing. Although classic red flags for hereditary cancer still apply, the difficulty arises in using these features to accurately distinguish truly sporadic (ie, non–germline mutated) cases of head and neck paragangliomas from single tumors that have a hereditary basis. Functional paragangliomas that seem to be sporadic have a ~12% rate of mutation detection rate when testing VHL , RET , SDHD , and SDHB , and nonsyndromic pheochromocytoma has a 24% mutation detection rate for RET , VHL , SDHD , and SDHB , with SDHD and SDHB as important contributors.
Regarding single head and neck paragangliomas, individuals with any hereditary features should undergo a genetic workup; that would include any individual with bilateral or multifocal head and neck paragangliomas, a single extra-adrenal paraganglioma in combination with a pheochromocytoma, or early-onset disease. Male gender can be considered an indication for testing. Sporadic head and neck paragangliomas occur more commonly in women, and a large study of head and neck paragangliomas found a 53% mutation detection rate for SDHD , SDHB , and SDHC testing in men. Malignant paraganglioma is also suggestive of a genetic cause, and the prevalence of an underlying SDHB mutation is not affect by a syndromic presentation or positive family history.
Some studies have demonstrated a high genetic contribution to seemingly sporadic extra-adrenal paragangliomas, indicating that there would be many missed cases. Parent-of-origin effects and reduced penetrance contribute to a low specificity of family history, which may be compounded by lack of family history information, unrecognized disease in relatives, and lifelong hereditary risk that extends into the diagnosis age range of sporadic tumors. A decade ago, common wisdom held that only about 10% of head and neck paragangliomas (outside the Netherlands, where the rate is much higher because of founder mutations) had a genetic cause, but soon after the identification of the SDH genes a significantly higher hereditary proportion was better appreciated. SDHD , SDHB , and SDHC mutations contribute to 9.5% of apparently sporadic, 71.4% of multiple cases, and 87.8% of familial head and neck paragangliomas. A genetic workup should thus be considered for most, if not all, head and neck paragangliomas and all malignant pheochromocytomas and extra-adrenal paragangliomas.
Differential Diagnosis
The VHL, MEN2, and NF1 syndromes each have anecdotal reports of head and neck paragangliomas. Paraganglioma, gastric stromal tumor, and pulmonary chondroma comprise the Carney triad, which has an unknown genetic basis. NF1 should be recognizable as a genodermatosis, and VHL and MEN2 are usually evident based on family history or clinical features. After exclusion of SDH genes, only about 0.5% of patients with head and neck paragangliomas have VHL mutations, whereas RET and NF1 mutations are rarer still, suggesting that VHL and RET testing could be foregone.
Patients with a single nonsyndromic pheochromocytoma or extra-adrenal paraganglioma can be stratified for testing (eg, SDH gene testing for head and neck paraganglioma); SDHB initially for thoracic-retroperitoneal paraganglioma, and VHL / SDHB / RET algorithms for pheochromocytoma. Renal cell carcinoma in combination with an extra-adrenal paraganglioma could suggest VHL, but, in the absence of other VHL features in the proband and family, SDHB testing could first be considered.
Diagnostics
DNA sequence analysis and deletion/duplication analysis are available on a clinical basis for SDHD , SDHB , and SDHC , but not SDHAF2 / SDH5 . The proportion of mutation types has been well summarized ; about 3% to 7% comprise large deletions that require analysis by multiplex ligation-dependent probe amplification (MLPA) or similar methods. Sensitivity of molecular genetic testing is probably about 70% to 90%. A study employing SDHB immunohistochemical (IHC) staining in 220 pheochromocytomas and paragangliomas showed that 6 tumors were SDHB IHC negative among 53 tumors without any detectable germline mutation, and 5 of the 6 cases had syndromic features. In the larger series, 102 tumors had detectable SDH mutations. This suggests a mutation detection rate of about 94% (102/108) for SDH gene testing using DNA sequencing and MLPA methodologies.
Because testing for numerous genes for head and neck paragangliomas is costly ($2700 for SDHD , SDHB , and SDHC in the United States); prioritization of genetic testing should be based on first-step predictors of positive family history, preceding pheochromocytoma, multiple tumors, malignant tumors, age 40 years or younger, and male gender which have a predicted sensitivity of 92% and predicted specificity of 94%. An approach to case stratification using IHC staining of SDHB in formalin-fixed paraffin-embedded pheochromocytomas and paragangliomas, analogous to tumor testing of mismatch repair proteins to identify Lynch syndrome, has also been presented.
Prognosis and Management
Treatment of head and neck paragangliomas is mainly surgical, but watchful waiting or radiation therapy may be the best approach for certain tumors such as slowly growing vagal and jugular fossa paragangliomas in older patients in whom surgery may be problematic. Because multicentricity is common, debility from cranial neuropathy is greatly magnified, thus all patients, including those with a single tumor, must undergo preoperative MRI of the head and neck, which also guides surgical planning. It is particularly important to spare at least 1 vagal nerve and its laryngeal branches. Although functional head and neck paragangliomas are rare, preoperative biochemical screening for catecholamines and metanephrines is strongly advised to avoid the catastrophic cardiovascular collapse that can be precipitated by surgery. Biochemical screening also assesses unrecognized or asymptomatic pheochromocytomas and functioning extra-adrenal paragangliomas that occur at significant rates with all of the SDH syndromes.
For patients with pheochromocytomas and extra-adrenal paragangliomas, preoperative achievement of β-adrenergic blockade and maintenance of adequate volume status, as well as intraoperative monitoring and surgical approaches, are detailed elsewhere. Surgical treatment of pheochromocytoma is usually curative, but follow-up testing is needed. Pheochromocytomas and extra-adrenal paragangliomas have an overall 5% to 15% rate of malignancy, but the risk of extra-adrenal paragangliomas and malignancy is higher in SDHB carriers. SDHB mutation has been found to be an independent predictor of mortality for malignant pheochromocytoma or extra-adrenal paraganglioma (relative risk 2.7). It has been suggested that SDHB testing be performed in all patients with malignant pheochromocytomas, and that carriers be more aggressively managed using a combination of surgery, metabolic radiotherapy (eg, 131 I-labeled m -iodobenzylguanidine or radioactive somatostatin analogues) with consideration of chemotherapy. Further details on diagnosis, localization, and treatment have been summarized elsewhere.
There has been no consensus conference outlining screening recommendations for the hereditary paraganglioma syndromes. One regimen is for head and neck MRI every 2 years beginning at age 16 years, along with urinary screening for pheochromocytoma every 2 years. Another recommends annual MRI of the neck, thorax, and abdomen/pelvis with [ 18F ]fluorodopa or [ 18F ]fluorodopamine positron emission tomography ( 18 F DOPA PET) as a possible alternative, coupled with screening for catecholamines and metanephrines. Scans can also be performed with 111 indium pentetreotide scintigraphy, a somatostatin analogue that binds to somatostatin type 2 receptors usually present in paragangliomas, coupled to a radioisotope. This method can detect small (5–10 mm) lesions with high accuracy. 18 F DOPA PET has higher sensitivity than MRI for lesions of less than 1 cm, and whole-body screening is a possible advantage, especially for SDHB carriers. The 3.4% prevalence of renal malignancy in SDHB opens the possibility for screening, particularly in families with this presentation. Age of onset may extend to less than 10 years, especially for SDHB . Genetic testing has been recommended to begin as early as age 5 to 10 years in SDH families, with annual screening in mutation carriers. The later age of onset seen across case series of SDHC carriers suggests that genetic testing and screening could be delayed.
Genetic Function and Genomic Imprinting
Mitochondrial complex II, the only complex in the respiratory chain completely encoded by nuclear genes, is a tetrameric protein encoded by 3 paraganglioma genes, SDHD , SDHB , and SDHC , and a fourth gene, SDHA , which is a cause of Leigh syndrome. The recently discovered paraganglioma gene, SDHAF2 / SDH5 , is required for flavination of the complex, as shown in yeast. Downregulation of SDH results in induction of the hypoxic pathway, and SDH has been proposed as an oxygen sensor, perhaps through the production of reactive oxygen species. PGL1 ( SDHD ) and, apparently, PGL2 ( SDHAF2 / SDH5 ) kindreds both show parent-of-origin effects, whereby carrier offspring of carrier fathers, but not carrier mothers, manifest disease with age-dependent penetrance liabilities. This observation was noted before linkage or cloning of the underlying genes and has held true with more direct evidence from mutational analysis, save for 1 reported exception.
The molecular basis of this parent-of-origin effect remains uncertain but models have been proposed. One possibility is that the SDHD gene is not imprinted, but that selective somatic loss of the maternal wild-type SDHD allele is driven by loss of gene(s) at 11p15 that undergo imprinting. This would constitute a single hit, leaving only paternally imprinted 11p15 gene(s) that are in phase with paternally inherited SDHD mutations, thereby mimicking SDHD imprinting. If PGL2 is more convincingly shown to have a parent-of-origin effect, the same mechanism could be used to explain this effect given its location on chromosome 11.
The role of these genes in neoplasia is also being studied. SDHD was the first tumor suppressor gene identified that encodes a protein involved in mitochondrial metabolism. A second example of a tumor suppressor gene with a role in mitochondrial metabolism is the fumarate hydratase ( FH ) gene. Cancer cells may gain a selective advantage by diverting glucose away from energy production and toward the production of macromolecules used to generate fatty acids, nonessential amino acids, and nucleotides (consistent with the Warburg effect first observed in 1924).
MEN1
History
Wermer reported the familial aggregation of pituitary tumors, multiple islet cell adenomas, diffuse parathyroid hyperplasia, and peptic ulcer disease, combinations of which had previously been reported only in individual patients. Separately, Zollinger and Ellison reported an association of primary peptic ulcerations of the jejunum and other unusual locations, unremitting gastric acid secretion, and non–insulin producing pancreatic islet cell tumors. Later, these clinical presentations were recognized as representing a spectrum of disease common to 1 syndrome, MEN1.
MEN1 is rare, occurring in up to 1 in 25,000 individuals based on autopsy data, although biochemical data suggest a prevalence closer to 10 to 175 per 100,000 individuals. Inheritance is autosomal dominant with equal sex distribution. MEN1 typically becomes clinically apparent in the 30s and 40s, usually as a result of symptomatic primary hyperparathyroidism. The syndrome is occasionally manifested in childhood and the diagnosis is sometimes delayed until later adulthood.
Clinical Characteristics
MEN1 is characterized by an autosomal dominant predisposition to develop endocrine tumors of the parathyroids, gastroenteropancreatic (GEP) tract, and pituitary, as well as carcinoid tumors, adrenocortical tumors, specific nonendocrine tumors, and particular skin manifestations. About 25 unique growths are associated with the syndrome, many of which are hormonally active, resulting in a plethora of clinical features. Parathyroid hyperplasia is the most common manifestation, and primary hyperparathyroidism is usually the heralding feature of MEN1. Primary hyperparathyroidism emerges in the early 20s, about 30 years earlier than sporadic hyperparathyroidism, and penetrance is 80% to 100% by age 40 years. Tumors of the anterior pituitary occur in 10% to 60% of cases, including prolactinomas (the most common), other functional growths secreting growth hormone, TSH, and adrenocorticotropic hormone (ACTH), as well as nonfunctional pituitary tumors.
GEP tumors are synonymous with neuroendocrine tumors, as per the WHO, and develop from endocrine cells in the pancreatic islets and the duodenal and gastric mucosa. Islet cell tumors are typically well differentiated and most are nonfunctioning tumors that nonetheless have malignant potential and pose crucial diagnostic and treatment dilemmas that are discussed later. Functioning islet cell tumors include gastrinomas (a cause of hypergastrinism, known as Zollinger-Ellison syndrome ), which affect about 50% of individuals; insulinomas, which affect 10% to 35% of MEN1 patients; glucaconomas; somatostatinomas; VIPomas; and GRFomas. Rarely, endocrine GEP tumors arise in other foregut locations including the first jejunal loop or biliary tract.
MEN1 carcinoid tumors (also neuroendocrine tumors as per the WHO classification ) arise mainly in the foregut. Most MEN1 carcinoids are nonfunctional and thus do not result in the carcinoid syndrome. Gastric carcinoids affect about 10% of MEN1 patients; bronchial and thymic carcinoids each affect about 2% of carriers. Adrenocortical tumors affect 20% to 40% of carriers but only rarely are functional. Malignant adrenocortical carcinomas are rare. Central nervous system tumors include meningiomas and, rarely, ependymomas.
Skin manifestations affect most MEN1 carriers and include facial angiofibromas, collagenomas, café au lait macules, as well as lipomas of the skin and viscera.
Neuroendocrine tumors have been distinguished from neuroendocrine carcinoma. The neuroendocrine tumors derive from the system of disseminated neuroendocrine cells, which have been termed clear cells or APUD cells (APUDomas), reflecting their underlying biochemistry of amine precursor uptake and decarboxylation, and many of these tumors are still referred to as carcinoids. Many, but not all, sporadic carcinoids produce serotonin, which leads to the carcinoid syndrome (flushing, diarrhea, valvular heart lesions, abdominal cramping, and other symptoms) and the noninterchangeability of these terms has led to some confusion. MEN1 carcinoids rarely produce the carcinoid syndrome.
Familial MEN1 is defined as at least 1 MEN1 case plus at least 1 first-degree relative with at least 1 of the 3 main endocrine manifestations. In the Netherlands, 5 categories are used: (1) parathyroid hyperplasia or adenomas; (2) neuroendocrine tumor of the pancreas (pancreatic endocrine tumor); (3) neuroendocrine tumor of the gastrointestinal tract, thymus, or bronchus; (4) pituitary abnormalities; and (5) adrenal hyperplasia or adenomas. In this schema, patients who meet at least 3 of 5 criteria have MEN1, and patients in an MEN1 family are affected if they have manifested at least 1 of the features.
Specific Clinical Manifestations of MEN1
The penetrance of hyperparathyroidism in MEN1 carriers, usually detected by increased calcium levels, is generally cited as approaching 100% by age 50 years. This has been interpreted to mean that lack of primary hyperparathyroidism by middle adulthood rules out a diagnosis of MEN1 and obviates further assessment. However, lower penetrance has been documented, for example, in a regional study that found hyperparathyroidism in only 73% of MEN1 patients by age 50 years and 83% by age 70 years.
Gastrinoma is diagnosed by high serum gastrin and increased gastric acid output. Patients may present with severe peptic ulcer disease and its complications. Penetrance is about 50% by age 50 years, and patients generally have multiple, small tumors with a high rate of metastasis. The classic presentation of glucagonoma includes skin rash, glucose intolerance, and hypoaminoacidemia. Insulinomas are associated with intractable hypoglycemia that can elicit a catecholamine response and cause neurologic symptoms.
Pituitary tumors
MEN1 pituitary tumors involve the anterior pituitary and are typically larger than sporadic pituitary tumors. Macroadenoma is seen in 85% of MEN1 cases, compared with 42% of sporadic cases. Multiple pituitary tumors can occur but are not common for MEN1. The usual age of onset for MEN1 pituitary disease is 30 to 35 years, similar to sporadic pituitary tumors, with a similar hormone profile (prolactin [62%], growth hormone [9%], ACTH [4%], cosecreting tumors most commonly with prolactin [10%], and nonsecreting tumors [15%]). The Burin variant of MEN1, seen in several Newfoundland Canadian families, includes a combination of primary hyperparathyroidism, high penetrance of prolactinoma, and low penetrance of gastrinoma.
Carcinoid tumors
Familial clustering of carcinoid tumors has been reported in some families, but germline MEN1 mutations are not evident with this presentation. Although most sporadic carcinoids arise in the midgut or hindgut, MEN1-associated carcinoids usually arise in the foregut, an atypical location that should prompt an MEN1 evaluation. In contrast to sporadic carcinoids, most MEN1 carcinoids are nonfunctional and are not associated with the carcinoid syndrome. The pathologic classification of most gastric carcinoids is type 2 gastric enterochromaffin–like cell carcinoids.
MEN1-related thymic carcinoids have a male predominance and account for about a quarter of all thymic carcinoids. In contrast, bronchial carcinoids have a female predominance. MEN1-related thymic carcinoids have a penetrance of about 2.6% by age 40 years, are insidious, are not associated with Cushing or carcinoid syndrome, can be detected with octreotide scanning, and seem to be more clinically aggressive than sporadic thymic carcinoids. There is evidence for clustering of thymic carcinoid in close relatives, and for an association between smoking and thoracic carcinoids.
Skin manifestations
Skin manifestations include collagenomas and multiple facial angiofibromas, similar to those seen in tuberous sclerosis but in different locations (ie, the upper lip and vermillion border of the lip), locations that are usually spared in tuberous sclerosis. Collagenomas (72%), café au lait macules (38%), lipomas (34%), confettilike hypopigmented macules (6%), and multiple gingival papules (6%) were seen in this series.
Differential Diagnosis
Primary hyperparathyroidism is seen in 2% to 4% of the general population. The genetic differential diagnosis of familial hyperparathyroidism includes MEN1, MEN2, familial isolated hyperparathyroidism, familial hypocalciuric hypercalcemia, and the hyperparathyroidism-jaw tumor syndrome. Multiglandular parathyroid involvement is typical of MEN1 and familial isolated hyperparathyroidism, whereas sporadic hyperparathyroidism typically exhibits parathyroid adenoma. Hyperparathyroidism is the most common presenting feature of MEN1 and occurs about 3 decades earlier than sporadic cases, at about age 20 to 25 years. However, diagnosis of hyperparathyroidism in MEN1 can be delayed, perhaps through inadequate family history taking, unfamiliarity with the syndrome, or splintered care among subspecialists.
MEN1 mutations are found in about 23% of familial isolated hyperparathyroidism, but the genetic basis in most families remains unexplained. The differential diagnosis also includes an overlap syndrome termed MEN4 (Online Mendelian Inheritance in Man [OMIM] 610755), involving pituitary adenomas with acromegaly and parathyroid hyperplasia caused by germline CDKN1B (p27Kip1) mutation. However this seems to be a rare cause of mutation-negative MEN1 families. About 20% to 30% of patients with the Zollinger-Ellison syndrome have MEN1. The percentage of patients with GRFomas who have MEN1 is high, about 33%, whereas patients with other pancreatic neuroendocrine tumors constitute less than 15%. Infertility may be part of the homozygous (or compound heterozygous) MEN1 state.
Genetics
MEN1 was mapped via genetic linkage studies to 11q13, with further narrowing of the candidate region using closely linked markers and haplotype analysis in affected families. No evidence of locus heterogeneity was found. Tumor tissue studies showing loss of heterozygosity at 11q13 in malignancies from MEN1 patients, such as insulinomas and parathyroid tumors, and in sporadic MEN1-associated tumors such as gastrinomas, provided additional mapping data. Murine and human comparative mapping analysis also contributed toward MEN1 gene localization. Further fine-scale mapping, including high-resolution radiation hybrid mapping, led to the ability to perform highly accurate predictive testing using linkage markers, opening the door for the first time to avoidance of invasive biochemical testing for noncarriers.
A precisely ordered transcript map for the 2.8-Mb region containing the MEN1 locus was created, providing 33 additional gene candidates. In 1997, Chandrasekharappa and colleagues reported the positional cloning of the MEN1 gene. Independent confirmation was provided by the European Consortium on MEN1. Additional large studies of unrelated MEN1 probands identified a variety of MENIN mutations in most, but not all, families with MEN1, a prevalence of truncating mutations consistent with tumor suppressor activity, commonly observed absence of germline mutations in familial hyperparathyroidism and familial acromegaly, occasional de novo germline mutations, and lack of genotype-phenotype correlations. A compilation of 1336 mutations reported as of 2008 confirmed a general lack of genotype-phenotype correlations (familial isolated hyperparathyroidism and the Burin MEN1 variant are notable exceptions), and reported that more than 70% are truncating mutations that are generally distributed across the genomic sequence.
Diagnostics
Molecular genetic testing for MEN1
Genetic testing is recommended in several situations : diagnostic testing in individuals who meet clinical criteria for sporadic or familial MEN1 or in individuals who do not meet formal criteria but are suspicious for MEN1, and presymptomatic or confirmatory testing in relatives of a known MEN1 mutation carrier. Presymptomatic carriers should undergo age-appropriate screening, whereas those without a familial mutation (and their children) can avoid expensive, potentially distressing lifelong screening. MEN1 genetic testing is appropriate in children because it clarifies whether screening is needed. Prenatal diagnosis is feasible in families with a known mutation.
Sensitivity of DNA sequencing is about 70% to 90% in probands with familial MEN1 syndrome. Analysis of gene deletions and gross rearrangements, detectable through gene dosage methods or Southern blot analysis, adds an additional sensitivity of about 1% to 3%. The rate of positive genetic test results falls off to about 65% in simplex cases without familial features, and is fewer still for those with single MEN1-related features. A clinical laboratory-based study found a ~70% rate of detectable mutations in cases with parathyroid and islet cell tumors (irrespective of family history). A lower mutation detection rate of 25% was found in cases with parathyroid and pituitary tumors (without pancreatic islet cell tumors); among 10 such sporadic cases (both tumors in a single individual; positive family history not reported), no mutations were found. The mutation detection rate was lowest for parathyroid-only cases. MEN1 mutations are identified in 20% to 25% of familial isolated hyperparathyroidism and seem to be more commonly associated with missense mutations than in classically affected MEN1 kindreds.
At least 10% of MEN1 cases are believed to arise de novo. Somatic mosaicism may explain the lower mutation detection rate seen in some index cases with multiple MEN1 tumors but no family history of MEN1. As in neurofibromatosis type 2, a germline mutation in de novo cases can be missed on leukocyte DNA analysis but, in theory, may be detectable in tumor analysis and in offspring.
Preoperative recognition of MEN1 significantly reduces the rate of surgical failures for parathyroidectomy. A preoperative 6-question panel was superior to chart-based clinical history for identifying such patients who were often young, male, and found to have parathyroid hyperplasia at the time of surgery. Prophylactic thymectomy can be planned at the same time only if the MEN1 syndrome is recognized preoperatively; however, not all authorities agree on the addition of this surgery to parathyroidectomy. A sustained successful postsurgical outcome requires astute recognition of the syndrome and prospective screening. A retrospective study of 1838 members of 34 MEN1 kindreds found that almost half of deaths in carriers were MEN1 related, chiefly from malignancies, occurring at an earlier median age of 47 years, with other risks of premature death documented in other studies, whereas a Finnish historical study showed no differences in age of death for obligate heterozygotes compared with controls. The effect of presymptomatic genetic diagnosis relies heavily on clinical interventions, which are continuously developing. Nonetheless, the effect of genetic screening in those without the familial mutation cannot be overemphasized, because those individuals (and their offspring) can forgo burdensome screening tests.
Screening
The systematic evaluation of asymptomatic children who carry MEN1 mutations reveals a high prevalence of MEN1 growths, including primary hyperparathyroidism, nonfunctioning pancreatic neuroendocrine tumors, and pituitary microadenomas. MEN1 carriers have occasionally been clinically affected as children; for example, a 5 year old with a pituitary macroadenoma secreting prolactin and growth hormone, insulinoma at age 5 years, parathyroid adenoma with hyperparathyroidism at age 8 years, and thymic NET at age 16 years. MEN1-related mortality in childhood is believed to be low, and screening is aimed at reducing morbidity. The age at which to initiate screening is based on earliest observation of the relevant disease manifestation. Annual biochemical testing with serum calcium and parathyroid hormone should be initiated by age 8 years. Beginning at age 5 years, individuals should undergo annual prolactin and insulin growth factor (IGF)-I testing, and head MRI every 3 years. Beginning at age 5 years, individuals should undergo fasting glucose and insulin levels. Beginning at age 20 years, testing should be done annually for chromogranin-A, glucagon, and proinsulin. Imaging should be done every 3 years using 111 In-diethylenetriaminepentaacetic acid (DTPA) octreotide scan, computerized tomography (CT), or MRI. Beginning at age 20 years, annual biochemical testing should include gastrin level measurements. If gastrin levels are high, gastric acid output should be measured. Secretin-stimulated gastrin testing is measured if gastrin or gastric acid output is high. No biochemical testing is available for early detection of carcinoid tumors. CT is recommended every 3 years to screen for foregut carcinoid. Octreotide scanning can detect thymic carcinoids and has been suggested for MEN1 carriers, along with chest CT or MRI. Clustering within families suggests that particular attention should be paid to screening in these kindreds.
The surgical approach to treating MEN1 primary hyperparathyroidism is subtotal parathyroidectomy or total parathyroidectomy with nondominant forearm autotransplantation of parathyroid tissue (or cryopreservation for subsequent autografting). Cervical thymectomy may be performed concurrently as a prophylactic surgery. The surgical management of gastrinoma has been controversial. Surgery for patients with sporadic and MEN1-associated Zollinger-Ellison syndrome can reduce the rate of hepatic metastasis, implying a survival benefit. There is now evidence that long-term survival in MEN1 patients with gastrinoma can be improved through surgery. Remaining controversies about the potential for cure and the role of the Whipple procedure are well described. Surgery is curative for most insulinomas, chiefly through enucleation but also distal pancreatectomy or pancreaticoduodenectomy for large or malignant tumors. Medical management of other functional, non–Zollinger-Ellison syndrome tumors is available (eg, somatostatin analogues such as octreotide and peptide receptor radionuclide therapy). These tumors merit surgical exploration and can frequently be cured. Prophylactic thymectomy during subtotal or total parathyroidectomy has been proposed as a way to reduce the risk of developing thymic carcinoid, particularly given its aggressive nature, and to reduce the risks of recurrent hyperparathyroidism following surgery. Avoidance of tobacco smoking and exposure to secondhand smoke is a practical approach given the suggestion that thymic carcinoid may be smoking related. There is no consensus on how to treat MEN1-related adrenal cortical tumors, but functional tumors should be resected.
Gene Function
The MEN1 gene has long been recognized as a tumor suppressor gene based on loss of the wild-type allele in MEN1-related tumors and the autosomal dominant tumor predisposition pattern seen in families. The MEN1 gene product, menin, is a nuclear protein with tumor suppressor functions and also plays a role in maintaining DNA stability and gene regulation. Menin has the unique property of promoting oncogenesis in the hematopoietic lineage but suppressing tumorigenesis in the endocrine lineage.
MEN1
History
Wermer reported the familial aggregation of pituitary tumors, multiple islet cell adenomas, diffuse parathyroid hyperplasia, and peptic ulcer disease, combinations of which had previously been reported only in individual patients. Separately, Zollinger and Ellison reported an association of primary peptic ulcerations of the jejunum and other unusual locations, unremitting gastric acid secretion, and non–insulin producing pancreatic islet cell tumors. Later, these clinical presentations were recognized as representing a spectrum of disease common to 1 syndrome, MEN1.
MEN1 is rare, occurring in up to 1 in 25,000 individuals based on autopsy data, although biochemical data suggest a prevalence closer to 10 to 175 per 100,000 individuals. Inheritance is autosomal dominant with equal sex distribution. MEN1 typically becomes clinically apparent in the 30s and 40s, usually as a result of symptomatic primary hyperparathyroidism. The syndrome is occasionally manifested in childhood and the diagnosis is sometimes delayed until later adulthood.
Clinical Characteristics
MEN1 is characterized by an autosomal dominant predisposition to develop endocrine tumors of the parathyroids, gastroenteropancreatic (GEP) tract, and pituitary, as well as carcinoid tumors, adrenocortical tumors, specific nonendocrine tumors, and particular skin manifestations. About 25 unique growths are associated with the syndrome, many of which are hormonally active, resulting in a plethora of clinical features. Parathyroid hyperplasia is the most common manifestation, and primary hyperparathyroidism is usually the heralding feature of MEN1. Primary hyperparathyroidism emerges in the early 20s, about 30 years earlier than sporadic hyperparathyroidism, and penetrance is 80% to 100% by age 40 years. Tumors of the anterior pituitary occur in 10% to 60% of cases, including prolactinomas (the most common), other functional growths secreting growth hormone, TSH, and adrenocorticotropic hormone (ACTH), as well as nonfunctional pituitary tumors.
GEP tumors are synonymous with neuroendocrine tumors, as per the WHO, and develop from endocrine cells in the pancreatic islets and the duodenal and gastric mucosa. Islet cell tumors are typically well differentiated and most are nonfunctioning tumors that nonetheless have malignant potential and pose crucial diagnostic and treatment dilemmas that are discussed later. Functioning islet cell tumors include gastrinomas (a cause of hypergastrinism, known as Zollinger-Ellison syndrome ), which affect about 50% of individuals; insulinomas, which affect 10% to 35% of MEN1 patients; glucaconomas; somatostatinomas; VIPomas; and GRFomas. Rarely, endocrine GEP tumors arise in other foregut locations including the first jejunal loop or biliary tract.
MEN1 carcinoid tumors (also neuroendocrine tumors as per the WHO classification ) arise mainly in the foregut. Most MEN1 carcinoids are nonfunctional and thus do not result in the carcinoid syndrome. Gastric carcinoids affect about 10% of MEN1 patients; bronchial and thymic carcinoids each affect about 2% of carriers. Adrenocortical tumors affect 20% to 40% of carriers but only rarely are functional. Malignant adrenocortical carcinomas are rare. Central nervous system tumors include meningiomas and, rarely, ependymomas.
Skin manifestations affect most MEN1 carriers and include facial angiofibromas, collagenomas, café au lait macules, as well as lipomas of the skin and viscera.
Neuroendocrine tumors have been distinguished from neuroendocrine carcinoma. The neuroendocrine tumors derive from the system of disseminated neuroendocrine cells, which have been termed clear cells or APUD cells (APUDomas), reflecting their underlying biochemistry of amine precursor uptake and decarboxylation, and many of these tumors are still referred to as carcinoids. Many, but not all, sporadic carcinoids produce serotonin, which leads to the carcinoid syndrome (flushing, diarrhea, valvular heart lesions, abdominal cramping, and other symptoms) and the noninterchangeability of these terms has led to some confusion. MEN1 carcinoids rarely produce the carcinoid syndrome.
Familial MEN1 is defined as at least 1 MEN1 case plus at least 1 first-degree relative with at least 1 of the 3 main endocrine manifestations. In the Netherlands, 5 categories are used: (1) parathyroid hyperplasia or adenomas; (2) neuroendocrine tumor of the pancreas (pancreatic endocrine tumor); (3) neuroendocrine tumor of the gastrointestinal tract, thymus, or bronchus; (4) pituitary abnormalities; and (5) adrenal hyperplasia or adenomas. In this schema, patients who meet at least 3 of 5 criteria have MEN1, and patients in an MEN1 family are affected if they have manifested at least 1 of the features.
Specific Clinical Manifestations of MEN1
The penetrance of hyperparathyroidism in MEN1 carriers, usually detected by increased calcium levels, is generally cited as approaching 100% by age 50 years. This has been interpreted to mean that lack of primary hyperparathyroidism by middle adulthood rules out a diagnosis of MEN1 and obviates further assessment. However, lower penetrance has been documented, for example, in a regional study that found hyperparathyroidism in only 73% of MEN1 patients by age 50 years and 83% by age 70 years.
Gastrinoma is diagnosed by high serum gastrin and increased gastric acid output. Patients may present with severe peptic ulcer disease and its complications. Penetrance is about 50% by age 50 years, and patients generally have multiple, small tumors with a high rate of metastasis. The classic presentation of glucagonoma includes skin rash, glucose intolerance, and hypoaminoacidemia. Insulinomas are associated with intractable hypoglycemia that can elicit a catecholamine response and cause neurologic symptoms.
Pituitary tumors
MEN1 pituitary tumors involve the anterior pituitary and are typically larger than sporadic pituitary tumors. Macroadenoma is seen in 85% of MEN1 cases, compared with 42% of sporadic cases. Multiple pituitary tumors can occur but are not common for MEN1. The usual age of onset for MEN1 pituitary disease is 30 to 35 years, similar to sporadic pituitary tumors, with a similar hormone profile (prolactin [62%], growth hormone [9%], ACTH [4%], cosecreting tumors most commonly with prolactin [10%], and nonsecreting tumors [15%]). The Burin variant of MEN1, seen in several Newfoundland Canadian families, includes a combination of primary hyperparathyroidism, high penetrance of prolactinoma, and low penetrance of gastrinoma.
Carcinoid tumors
Familial clustering of carcinoid tumors has been reported in some families, but germline MEN1 mutations are not evident with this presentation. Although most sporadic carcinoids arise in the midgut or hindgut, MEN1-associated carcinoids usually arise in the foregut, an atypical location that should prompt an MEN1 evaluation. In contrast to sporadic carcinoids, most MEN1 carcinoids are nonfunctional and are not associated with the carcinoid syndrome. The pathologic classification of most gastric carcinoids is type 2 gastric enterochromaffin–like cell carcinoids.
MEN1-related thymic carcinoids have a male predominance and account for about a quarter of all thymic carcinoids. In contrast, bronchial carcinoids have a female predominance. MEN1-related thymic carcinoids have a penetrance of about 2.6% by age 40 years, are insidious, are not associated with Cushing or carcinoid syndrome, can be detected with octreotide scanning, and seem to be more clinically aggressive than sporadic thymic carcinoids. There is evidence for clustering of thymic carcinoid in close relatives, and for an association between smoking and thoracic carcinoids.
Skin manifestations
Skin manifestations include collagenomas and multiple facial angiofibromas, similar to those seen in tuberous sclerosis but in different locations (ie, the upper lip and vermillion border of the lip), locations that are usually spared in tuberous sclerosis. Collagenomas (72%), café au lait macules (38%), lipomas (34%), confettilike hypopigmented macules (6%), and multiple gingival papules (6%) were seen in this series.
Differential Diagnosis
Primary hyperparathyroidism is seen in 2% to 4% of the general population. The genetic differential diagnosis of familial hyperparathyroidism includes MEN1, MEN2, familial isolated hyperparathyroidism, familial hypocalciuric hypercalcemia, and the hyperparathyroidism-jaw tumor syndrome. Multiglandular parathyroid involvement is typical of MEN1 and familial isolated hyperparathyroidism, whereas sporadic hyperparathyroidism typically exhibits parathyroid adenoma. Hyperparathyroidism is the most common presenting feature of MEN1 and occurs about 3 decades earlier than sporadic cases, at about age 20 to 25 years. However, diagnosis of hyperparathyroidism in MEN1 can be delayed, perhaps through inadequate family history taking, unfamiliarity with the syndrome, or splintered care among subspecialists.
MEN1 mutations are found in about 23% of familial isolated hyperparathyroidism, but the genetic basis in most families remains unexplained. The differential diagnosis also includes an overlap syndrome termed MEN4 (Online Mendelian Inheritance in Man [OMIM] 610755), involving pituitary adenomas with acromegaly and parathyroid hyperplasia caused by germline CDKN1B (p27Kip1) mutation. However this seems to be a rare cause of mutation-negative MEN1 families. About 20% to 30% of patients with the Zollinger-Ellison syndrome have MEN1. The percentage of patients with GRFomas who have MEN1 is high, about 33%, whereas patients with other pancreatic neuroendocrine tumors constitute less than 15%. Infertility may be part of the homozygous (or compound heterozygous) MEN1 state.
Genetics
MEN1 was mapped via genetic linkage studies to 11q13, with further narrowing of the candidate region using closely linked markers and haplotype analysis in affected families. No evidence of locus heterogeneity was found. Tumor tissue studies showing loss of heterozygosity at 11q13 in malignancies from MEN1 patients, such as insulinomas and parathyroid tumors, and in sporadic MEN1-associated tumors such as gastrinomas, provided additional mapping data. Murine and human comparative mapping analysis also contributed toward MEN1 gene localization. Further fine-scale mapping, including high-resolution radiation hybrid mapping, led to the ability to perform highly accurate predictive testing using linkage markers, opening the door for the first time to avoidance of invasive biochemical testing for noncarriers.
A precisely ordered transcript map for the 2.8-Mb region containing the MEN1 locus was created, providing 33 additional gene candidates. In 1997, Chandrasekharappa and colleagues reported the positional cloning of the MEN1 gene. Independent confirmation was provided by the European Consortium on MEN1. Additional large studies of unrelated MEN1 probands identified a variety of MENIN mutations in most, but not all, families with MEN1, a prevalence of truncating mutations consistent with tumor suppressor activity, commonly observed absence of germline mutations in familial hyperparathyroidism and familial acromegaly, occasional de novo germline mutations, and lack of genotype-phenotype correlations. A compilation of 1336 mutations reported as of 2008 confirmed a general lack of genotype-phenotype correlations (familial isolated hyperparathyroidism and the Burin MEN1 variant are notable exceptions), and reported that more than 70% are truncating mutations that are generally distributed across the genomic sequence.
Diagnostics
Molecular genetic testing for MEN1
Genetic testing is recommended in several situations : diagnostic testing in individuals who meet clinical criteria for sporadic or familial MEN1 or in individuals who do not meet formal criteria but are suspicious for MEN1, and presymptomatic or confirmatory testing in relatives of a known MEN1 mutation carrier. Presymptomatic carriers should undergo age-appropriate screening, whereas those without a familial mutation (and their children) can avoid expensive, potentially distressing lifelong screening. MEN1 genetic testing is appropriate in children because it clarifies whether screening is needed. Prenatal diagnosis is feasible in families with a known mutation.
Sensitivity of DNA sequencing is about 70% to 90% in probands with familial MEN1 syndrome. Analysis of gene deletions and gross rearrangements, detectable through gene dosage methods or Southern blot analysis, adds an additional sensitivity of about 1% to 3%. The rate of positive genetic test results falls off to about 65% in simplex cases without familial features, and is fewer still for those with single MEN1-related features. A clinical laboratory-based study found a ~70% rate of detectable mutations in cases with parathyroid and islet cell tumors (irrespective of family history). A lower mutation detection rate of 25% was found in cases with parathyroid and pituitary tumors (without pancreatic islet cell tumors); among 10 such sporadic cases (both tumors in a single individual; positive family history not reported), no mutations were found. The mutation detection rate was lowest for parathyroid-only cases. MEN1 mutations are identified in 20% to 25% of familial isolated hyperparathyroidism and seem to be more commonly associated with missense mutations than in classically affected MEN1 kindreds.
At least 10% of MEN1 cases are believed to arise de novo. Somatic mosaicism may explain the lower mutation detection rate seen in some index cases with multiple MEN1 tumors but no family history of MEN1. As in neurofibromatosis type 2, a germline mutation in de novo cases can be missed on leukocyte DNA analysis but, in theory, may be detectable in tumor analysis and in offspring.
Preoperative recognition of MEN1 significantly reduces the rate of surgical failures for parathyroidectomy. A preoperative 6-question panel was superior to chart-based clinical history for identifying such patients who were often young, male, and found to have parathyroid hyperplasia at the time of surgery. Prophylactic thymectomy can be planned at the same time only if the MEN1 syndrome is recognized preoperatively; however, not all authorities agree on the addition of this surgery to parathyroidectomy. A sustained successful postsurgical outcome requires astute recognition of the syndrome and prospective screening. A retrospective study of 1838 members of 34 MEN1 kindreds found that almost half of deaths in carriers were MEN1 related, chiefly from malignancies, occurring at an earlier median age of 47 years, with other risks of premature death documented in other studies, whereas a Finnish historical study showed no differences in age of death for obligate heterozygotes compared with controls. The effect of presymptomatic genetic diagnosis relies heavily on clinical interventions, which are continuously developing. Nonetheless, the effect of genetic screening in those without the familial mutation cannot be overemphasized, because those individuals (and their offspring) can forgo burdensome screening tests.
Screening
The systematic evaluation of asymptomatic children who carry MEN1 mutations reveals a high prevalence of MEN1 growths, including primary hyperparathyroidism, nonfunctioning pancreatic neuroendocrine tumors, and pituitary microadenomas. MEN1 carriers have occasionally been clinically affected as children; for example, a 5 year old with a pituitary macroadenoma secreting prolactin and growth hormone, insulinoma at age 5 years, parathyroid adenoma with hyperparathyroidism at age 8 years, and thymic NET at age 16 years. MEN1-related mortality in childhood is believed to be low, and screening is aimed at reducing morbidity. The age at which to initiate screening is based on earliest observation of the relevant disease manifestation. Annual biochemical testing with serum calcium and parathyroid hormone should be initiated by age 8 years. Beginning at age 5 years, individuals should undergo annual prolactin and insulin growth factor (IGF)-I testing, and head MRI every 3 years. Beginning at age 5 years, individuals should undergo fasting glucose and insulin levels. Beginning at age 20 years, testing should be done annually for chromogranin-A, glucagon, and proinsulin. Imaging should be done every 3 years using 111 In-diethylenetriaminepentaacetic acid (DTPA) octreotide scan, computerized tomography (CT), or MRI. Beginning at age 20 years, annual biochemical testing should include gastrin level measurements. If gastrin levels are high, gastric acid output should be measured. Secretin-stimulated gastrin testing is measured if gastrin or gastric acid output is high. No biochemical testing is available for early detection of carcinoid tumors. CT is recommended every 3 years to screen for foregut carcinoid. Octreotide scanning can detect thymic carcinoids and has been suggested for MEN1 carriers, along with chest CT or MRI. Clustering within families suggests that particular attention should be paid to screening in these kindreds.
The surgical approach to treating MEN1 primary hyperparathyroidism is subtotal parathyroidectomy or total parathyroidectomy with nondominant forearm autotransplantation of parathyroid tissue (or cryopreservation for subsequent autografting). Cervical thymectomy may be performed concurrently as a prophylactic surgery. The surgical management of gastrinoma has been controversial. Surgery for patients with sporadic and MEN1-associated Zollinger-Ellison syndrome can reduce the rate of hepatic metastasis, implying a survival benefit. There is now evidence that long-term survival in MEN1 patients with gastrinoma can be improved through surgery. Remaining controversies about the potential for cure and the role of the Whipple procedure are well described. Surgery is curative for most insulinomas, chiefly through enucleation but also distal pancreatectomy or pancreaticoduodenectomy for large or malignant tumors. Medical management of other functional, non–Zollinger-Ellison syndrome tumors is available (eg, somatostatin analogues such as octreotide and peptide receptor radionuclide therapy). These tumors merit surgical exploration and can frequently be cured. Prophylactic thymectomy during subtotal or total parathyroidectomy has been proposed as a way to reduce the risk of developing thymic carcinoid, particularly given its aggressive nature, and to reduce the risks of recurrent hyperparathyroidism following surgery. Avoidance of tobacco smoking and exposure to secondhand smoke is a practical approach given the suggestion that thymic carcinoid may be smoking related. There is no consensus on how to treat MEN1-related adrenal cortical tumors, but functional tumors should be resected.
Gene Function
The MEN1 gene has long been recognized as a tumor suppressor gene based on loss of the wild-type allele in MEN1-related tumors and the autosomal dominant tumor predisposition pattern seen in families. The MEN1 gene product, menin, is a nuclear protein with tumor suppressor functions and also plays a role in maintaining DNA stability and gene regulation. Menin has the unique property of promoting oncogenesis in the hematopoietic lineage but suppressing tumorigenesis in the endocrine lineage.
Related posts:
 Clinical Genetics of Hereditary Colorectal Cancer
Hereditary Genodermatoses with Cancer Predisposition
Genitourinary Cancer Predisposition Syndromes
Hereditary Genodermatoses with Cancer Predisposition
Genitourinary Cancer Predisposition Syndromes
Endocrine Cancer Predisposition Syndromes: Hereditary Paraganglioma, Multiple Endocrine Neoplasia Type 1, Multiple Endocrine Neoplasia Type 2, and Hereditary Thyroid Cancer
Clinical Genetics of Hereditary Colorectal Cancer
Hereditary Genodermatoses with Cancer Predisposition
Genitourinary Cancer Predisposition Syndromes
Hereditary Genodermatoses with Cancer Predisposition
Genitourinary Cancer Predisposition Syndromes
Endocrine Cancer Predisposition Syndromes: Hereditary Paraganglioma, Multiple Endocrine Neoplasia Type 1, Multiple Endocrine Neoplasia Type 2, and Hereditary Thyroid Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


