
The investigation of these disorders has always provided a unique window to understand basic biology and disease pathophysiology. Yet these molecular and biologic discoveries have had limited clinical impact on the hemoglobinopathy population. Supportive care, transfusion therapy, and hydroxyurea remain the only available therapies for most patients, and these options have not changed in decades. As a clinician and investigator, I am optimistic that the accomplishments described within this issue will result in decreased morbidity and improved quality of life for the patient population. However, it is important to view these discoveries in a historical context to understand the scientific chain of events that led us to these advances and the disappointment of earlier, promising therapeutic discoveries that did not achieve efficacy for the medical and patient community.
Historically, clinical symptoms of hemoglobinopathies appear to be mentioned in Hippocratic writings and have long been observed in African tribal histories. Centuries ago, the diseases were given onomatopoeic names that indicate severe pain and suffering. The African medical literature of the 1870s referred to a disease called “ogbanjes” (“children who come and go”) because of the high infant mortality rate with this syndrome. In 1910, Drs Herrick and Irons described the sickle cell erythrocyte in the peripheral smear of Dr Noel, a dental student from Granada. This was rapidly followed by the work of Emmel and Hahn, who demonstrated that sickling was an oxygen-dependent phenomenon. In 1934, Drs Diggs and Ching concluded that the symptoms of sickle cell disease were caused by occlusion of the microvasculature. Sherman, in 1940, noted that polymerization was a key factor in the sickling process and secondary vascular obstruction. A decade later Singer and Singer observed this polymerization was inhibited in the presence of fetal hemoglobin. This was followed by Hofrichter and Eaton, who found that the delay time for the initiation of deoxy-hemoglobin-S polymerization was dependent on hemoglobin S concentration and the transit time of red cells.
Sickle cell was classified as the first molecular disease by Pauling and colleagues in 1949, who concluded from their electrophoretic studies that the basic pathology was due to a change in the amino acid structure of hemoglobin. Ingram then identified that sickle hemoglobin differed from normal hemoglobin by a single amino acid substitution of valine for glutamic acid, utilizing the newly developed fingerprinting technique to identify amino acid changes. In the early 1970s, recombinant DNA technology resulted in nucleic acid sequencing showing there was an A → T DNA change in the sickle hemoglobin DNA, which changes the glutamine codon to valine.
In 1980, the understanding that sickle cell disease is a complex vasculopathy was initiated by the seminal work of Hebbel and colleagues, who wrote that sickled red cells abnormally adhere to endothelium and correlate with disease severity. These observations led to the central role of ischemia reperfusion injury and inflammation in sickle cell disease pathology. In 2002, Reiter and colleagues noted that nitric oxide depletion in sickle cell disease is induced by free hemoglobin and amplifies the vasculopathy.
Critical advances in the pathophysiology of thalassemia and iron regulation were occurring concomitantly to discoveries in sickle cell disease. Weatherall and colleagues utilized quantitative methods of hemoglobin synthesis to determine that defective and imbalanced globin synthesis were an essential part of the pathophysiology in the thalassemia syndromes. Finch and Sturgeon, in studying iron and erythrokinetics in patients, demonstrated that marked ineffective erythropoiesis is a hallmark of thalassemia and is responsible for marrow expansion and increased iron absorption regardless of body iron stores. In 1978, Hershko and colleagues described the toxic non-transferrin-bound fraction of plasma iron, which leads to iron-induced free radical damage. In 2001, Park and colleagues discovered the peptide hepcidin , which drives the increased gastrointestinal iron absorption in thalassemia.
In the last 50 years, several promising therapies have been proposed based on biologic discoveries. In the 1970s, anti-sickling agents designed to impair polymerization by altering hemoglobin ligands entered clinical trials. Urea , initially claimed to decrease sickling, was ineffective in clinical trials and may have increased morbidity. Cyanate , which appeared to increase oxygen affinity, resulted in severe neurologic toxicity. Many other anti-sickling agents designed to alter noncovalent and covalent ligands were clinically ineffective. These were followed by clinical trials with membrane-active drugs designed to increase cell hydration. Initial studies with the antidiuretic hormone DDAVP were encouraging, but were discontinued due to lack of efficacy and toxicity. Cetiedil induced significant improvement in red cell deformability and hydration by inhibiting the Gardos pathway. Initial clinical observations were positive, but definitive pharmaceutical trials were never undertaken. Several other membrane-active agents were initiated. A novel Gardos channel inhibitor, senicapoc, significantly increased hemoglobin levels and improved red cell deformability, but increased pain in phase III trials resulted in its abrupt abandonment. The results of these trials support the theory of different subphenotypes of sickle cell disease with the possibility of drug-induced phenotypic shifts. Agents that alter red cell rheology and vascular flow entered clinical studies in the early 1980s. Pluronic F-68 (poloxamer 188), a nonionic copolymer surfactant, showed promise in phase II trials for acute pain but was disappointing in phase III trials, with a benefit seen only in a subpopulation of children—again suggesting that drug response may be not be the same in all populations.
The only successful FDA-approved pharmacologic approach to sickling has been in fetal hemoglobin induction. Following the seminal observations by DeSimone and colleagues that 5-azacytidine therapy increases hemoglobin F, multiple clinical trials were undertaken with different agents. 5-Azacytidine increased hemoglobin in both thalassemia and sickle cell patients. Its utilization was abandoned because of mutagenic and carcinogenic risks. Initially, it was thought to activate hemoglobin F synthesis by increased gamma gene expression by affecting methylation. However, several studies with multiple S-phase cytoxic drugs showed a similar hemoglobin F response. It was concluded that hemoglobin F was indirectly increased due to alteration of erythroid progenitor kinetics. Hydroxyurea , the least toxic, orally available agent underwent the most clinical studies, leading to its eventual FDA approval for use in adults with sickle cell disease. Exciting work with butyrate and other histone deacetylase inhibitors in the mid-1980s suggested they would be efficacious. However, clinical trials were less encouraging possibly due to drug metabolism and/or inhibition of erythropoiesis. Stimulation of fetal hemoglobin synthesis by erythropoietin has been demonstrated for decades, but the modest response and potential toxicity have prevented larger trials.
The biologic advances in sickle cell disease and therapeutic options described in this issue grew out of the scientific foundation described above. The first two articles give an overview of the evolving understanding of the pathophysiology. Dr Kuypers discusses the red cell membrane changes—including increased phosphatidyl serine (PS) exposure—that result in the sickle cell initiating a red cell vasculopathy. He briefly discusses potential therapies to address membrane alterations such as d -annexin binding to PS surfaces. Dr Hebbel provides an insightful discussion of how ischemia reperfusion injury is most likely responsible for much of the clinical syndrome of sickle cell disease, and he reviews the therapeutic options for ischemic reperfusion injury.
Therapeutic interventions can attack sickle cell pathology from the primary mutation to its downstream effects such as inflammation and thrombosis. Drs Chandrakasan and Malik discuss the advances in gene transfer vector technology and self-inactivating lentivirus that have resulted in the initial pilot success in thalassemia. They review recent discoveries that will improve clinical options, including gene editing technology, newer vectors, and induced pleuripotent stem cells.
The re-emergence of research interest in the causal relationship between decreased oxygen affinity of hemoglobin S and increased polymerization has resulted in promising pilot clinical studies with hemoglobin modifiers that increase oxygen affinity. Dr Safo and Dr Kato review the pathophysiologic effects resulting from sickle red cells exposed to low oxygen affinity and relevant therapeutic approaches, including the investigations with the aldehyde Aes-1035-HMF.
For several decades, increasing fetal hemoglobin has been a major focus in the treatment of hemoglobinopathies. Except for hydroxyurea, which received FDA approval 20 years ago, there remains no other commercially available hemoglobin F inducer. Drs Perrine, Pace, and Fowler review the history of hemoglobin F therapeutics and the recent advances in the understanding of fetal hemoglobin regulation. They discuss the new generation of noncytotoxic hemoglobin F inducers, including short-chain fatty acids, low-dose decitabine tetrahydrouridine, benserazide, as well as combinations with epigenetic modifiers and agents that increase translation. Dr Fibach and Dr Rachmilewitz review the effects of erythropoietin administration on hemoglobin F production as well as erythropoiesis, iron utilization, and oxidative stress. They discuss potential trials of erythropoietin alone or in combination with new emerging therapies.
Sickle cell disease is now considered an acute and chronic inflammatory condition. Dr Hoppe summarizes the in vitro and in vivo studies indicating the importance of inflammation in the development of its vasculopathy. Her article provides an overview of the cellular mechanisms involved in the inflammatory response and therapeutic agents that target these pathways. Discussion of key pathways involving adenosine signaling, nitric oxide/arginine dysregulation, selectins, and coagulation is expanded in individual articles. Drs Field, Nathan, and Linden review the important beneficial and detrimental effects of adenosine signaling in SCD pathophysiology. They discuss the anti-inflammatory effect of adenosine resulting from decreased activation of invariant NKT cells mediated by the adenosine A 2A receptor and the proinflammatory response to binding with the adenosine A 2B receptor. The in vitro and clinical studies with adenosine therapeutics are reviewed, including A 2A R agonist regadenoson, and A 2B R antagonist pegylated -ADA. Dr Morris reviews the recent studies that demonstrate arginine metabolomics and the dysregulation of nitric oxide hemostasis that plays an important role in the pathophysiology of sickle cell disease. The clinical benefit of arginine supplementation alone or with hydroxyurea in the treatment of leg ulcers, pulmonary hypertension, priapism, and pain is discussed.
Vaso-occlusion and impaired microvascular flow characterize the pathophysiology of sickle cell disease. The adhesive interactions involving erythrocytes, the endothelium, leukocytes, and platelets are central to this process. Selectins are adhesive receptors expressed on these cells that initiate the adhesive interactions. There are multiple species of selectins involved in this process. Drs Kultar and Embury discuss the key role of endothelial P-selectin-mediated cell adhesion as an initiator of abnormal blood flow in sickle cell disease. The authors reviewed the beneficial effects observed in pilot studies with P-selectin blocking agents, including pentosan polysulfate sodium. Dr Telen provides an informative review of the importance of E- and L-selectin in sickle cell disease pathology. GMI1070, a small carbohydrate molecule that binds to selectins—particularly E-selectin, is discussed in detail with highlights of the successful early trials. Dr Pakbaz and Dr Wun highlight the contribution of hemostatic abnormalities in the process of vaso-occlusion and impaired microvascular flow. They review the data showing increased risk of thromboembolic events, the potential role of platelet inhibitors, and anticoagulants.
The issue concludes with recent advances in understanding pathways that modulate erythropoiesis and iron physiology. Drs Rivella and Breda discuss the important role that JAK2 activity has in stress erythropoiesis and the potential therapeutic benefits of JAK2 inhibitors. They discuss the effects of activin signaling in hematopoiesis and the therapeutic strategies targeting deregulation of activin signaling—including clinical studies with ACE/RAP-011 and ACE536. Drs Schmidt and Fleming review the advances in understanding the hepcidin-ferroportin iron regulatory axis. They discuss new approaches to treating iron overload diseases utilizing hepcidin mimetics or by altering endogenous heparin expression. They highlight lipid nanoparticle encapsulated siRNA and antisense oligonucleotide ASO-mediated inhibition of TMPRSS6, and mini-hepcidin therapy. In β-thalassemia intermedia, they discuss the murine model results with transferrin therapy and hepcidin up-regulation.
Related posts:
 Gene Therapy for Hemoglobinopathies
Therapeutic Strategies to Alter the Oxygen Affinity of Sickle Hemoglobin
Gene Therapy for Hemoglobinopathies
Therapeutic Strategies to Alter the Oxygen Affinity of Sickle Hemoglobin
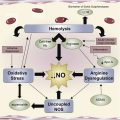 Alterations of the Arginine Metabolome in Sickle Cell Disease
Alterations of the Arginine Metabolome in Sickle Cell Disease
 Ischemia-reperfusion Injury in Sickle Cell Anemia
The Role of Adenosine Signaling in Sickle Cell Therapeutics
Ischemia-reperfusion Injury in Sickle Cell Anemia
The Role of Adenosine Signaling in Sickle Cell Therapeutics
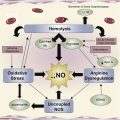 Alterations of the Arginine Metabolome in Sickle Cell Disease
Alterations of the Arginine Metabolome in Sickle Cell Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


