Fig. 11.1
5-FU-dependent depletion of MDSCs. 5-FU specifically targets and depletes MDSCs, reducing the overall immunosuppression. However, 5-FU treatment also induces a permeabilization of the lysosome in MDSCs. Cathepsin B can then escape the lysosome and enter the cytoplasm where it interacts with NLRP3, inducing the formation and activation of the NLRP3 inflammasome. The activated NLRP3 inflammasome cleaves the pro-IL-1β into active IL-1β that activates CD4 T cells to produce more IL-17, enhancing neoangiogenesis and tumor growth
The impact of chemotherapies on MDSCs is often a double-edged sword, tumor cells and immunosuppressive immune cells being masters at finding a loophole allowing for the return of an immunosuppressive tumor environment as seen here. We should be careful not to make any precocious conclusion before the full picture is before our eyes.
11.3 Effect of Chemotherapies on MDSCs in Human
Studies in humans on the effect of chemotherapies on MSDCs have sometimes shown contradictory data. In patients with a pancreatic cancer, 5-FU and gemcitabine were first shown to reduce MDSC percentage in about 40% of patients and in most of patients when associated with the GV101 vaccine using GM-CSF as an adjuvant. MDSC number did however come up in some patients, and this was correlated with an increase of pro-inflammatory cytokines [28]. These results match with a more recent study where treatment with gemcitabine or 5-FU was associated with an upregulation of GM-CSF secreted by tumor cells inducing the differentiation of monocytes in MDSCs [29]. On the contrary, a positive effect of gemcitabine and 5-FU on MDSCs was observed when associated with an immunotherapy consisting in cytokine-induced killer cells [30]. In metastatic renal cell carcinoma and pancreatic cancer, the use of this chemotherapy in association with an immunotherapy successfully reduced the number of MDSCs in the peripheral blood of patients and increased the survival time. Interestingly, in colorectal cancer patients, the positive or negative outcome of a 5-FU treatment is dependent on the type of combination used in association with 5-FU. Indeed, 5-FU is not used alone in colorectal cancer. The use of 5-FU with folic acid and oxaliplatin (FOLFOX) was proven beneficial with a decrease of the overall immunosuppression and MDSC percentage, whereas the association of 5-FU with folic acid and CPT11 (FOLFIRI) was detrimental and even increased the number of MDSCs in patients [31]. In another study testing the effect of FOLFOX associated with bevacizumab, an anti-VEGF-A antibody, on MDSCs in patients treated in first line of metastatic colorectal cancer, authors observed a drop of PMN-MDSCs in 15 out of 25 patients [32]. As cancer chemotherapies always associate several agents, it is crucial to study the effects of these associations.
11.4 Tyrosine Kinase Inhibitors
Aside from cytotoxic chemotherapies, several other classes of anticancer agents have been studied for their capability to block MDSC proliferation or to enhance their differentiation. Tyrosine kinase inhibitors (TKIs) are a group of molecules aiming to target tyrosine kinases in various pathways. Targeted pathways, the RAS-RAF/MAPK pathway, the PI3K/AKT/mTOR pathway, and the EGFR pathway, are involved in the regulation of cell survival, proliferation, differentiation, migration, and angiogenesis [33, 34]. Mutations in these pathways are often found in cancer, explaining the rapid and ongoing development of TKI these past few years [35, 36]. The potential effects of TKIs on MDSCs have raised a growing interest.
Sunitinib is a TKI targeting multiple receptor tyrosine kinases including VEGF-R1 and VEGF-R2, PDGF-Rs, but also c-KIT. It was approved by the FDA for the treatment of advanced renal cell carcinoma (RCC) in 2007 and is currently used in the frontline treatment for RCC. In RCC, sunitinib reversed MDSC accumulation by affecting their viability and proliferation. The decrease in MDSCs was linked to an increase in IFN-gamma production by CD3 cells [37]. Sunitinib decreased the number of MDSCs and Tregs as well as the production of the immunosuppressive cytokines IL-10, TGF-beta. Interestingly, the expression of the negative costimulatory molecules CTLA-4 and PD-1 on CD4 and CD8 T cells was decreased after a sunitinib treatment [38]. Sunitinib may reduce the expansion of monocytic MDSC while inducing apoptosis in the granulocytic MDSC subset [39]. However, intratumoral MDSC number and function were not affected by sunitinib, as the high quantity of GM-CSF produced in the tumor bed was protecting MDSCs in a STAT5-dependent pathway [40, 41]. Sunitinib was reported to also affect other cell types than MDSCs as reduction in the percentage of neutrophils and monocytes and an increase in lymphoid cells can be observed [37] (Fig. 11.2).
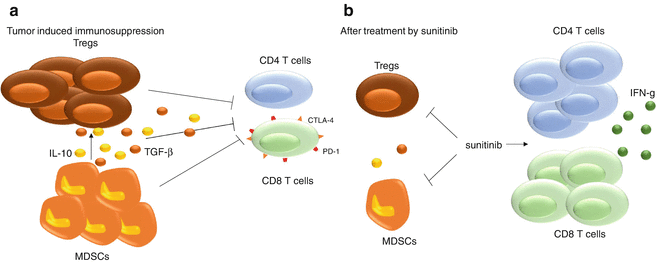
Fig. 11.2
Sunitinib immune effects. Sunitinib, a TKI targeting VEGF-Rs and PDGF-Rs, blocks MDSC accumulation by reducing their viability and proliferation. Sunitinib also targets Tregs and decreases IL-10 and TGF-β production while enhancing the proliferation of CD4 and CD8 T cells along with increasing the production of IFN-γ
A study using another VEGF pathway inhibitor, bevacizumab, an anti-VEGF-A mAb, showed that MDSCs were responsible for the refractoriness to clinical effect of anti-VEGF therapy [42], but no effect of bevacizumab on MDSC viability or differentiation was observed. This was later confirmed by another study showing that bevacizumab treatment did not decrease the percentage of MDSCs nor change their level of arginase-1 expression [43]. However, in patients with non-small cell lung carcinoma, three cycles of bevacizumab associated with chemotherapy regimens could reduce PMN-MDSC numbers in a bevacizumab-dependent way [44]. The impact of bevacizumab on MDSCs remains to be confirmed, and observed difference may be consequences of additional drugs used to treat cancer or due to the tumor types.
Sorafenib is an inhibitor directed against several kinases, among which are C-RAF, BRAF, and VEGF-R2 and VEGF-R3. Sorafenib was first demonstrated to have the capability to reduce Tregs and MDSCs in a murine liver cancer model, along with a slower tumor growth [45]. In addition, sorafenib was able to decrease the suppressive activity of MDSCs on CD8 T cells, while sunitinib, another inhibitor, could not [41]. Different protocols of administration with various doses were tested, and repetitive low doses of sorafenib appeared to enhance the efficacy of adoptive T cell therapy by decreasing MDSCs and Tregs but also by decreasing the expression of immunosuppressive molecules like IL-10 or TGF-beta [46]. Along with the selected dosage, the kinetic of treatment should also be considered as sorafenib could reduce the percentage of MDSC derived from monocytes but did not affect already differentiated MDSCs. Sorafenib effects might however not be restricted to MDSCs. Indeed, sorafenib decreased STAT1 and STAT5 phosphorylation in T cells, B cells, NK cells, Tregs, and MDSCs after stimulation with IL-2 or IFN-alpha [47]. Such data suggest that sorafenib could have deleterious effect on effector cells of the adaptive immune response. We probably should keep in mind these data when using sorafenib to deplete MDSCs.
Specific for c-kit and BCR-ABL, imatinib was the first TKI approved for chronic myeloid leukemia [48]. Imatinib efficiently reduced MDSC expansion and arginase-1 expression [49]. However, various reports also mention contradictory results in regard to its effects on other immune cell populations. Imatinib was shown to impair Tregs immunosuppressive functions [50], restore plasmacytoid dendritic cell function, and suppress tumor-induced CD4+ T cell tolerance [51, 52]. On the other hand, imatinib treatment was also shown to block the expansion of antigen-experienced CD8 T cells while leaving primary T and B cell responses unaffected [53]. Dasatinib is a second-generation compound used in patients with chronic myeloid leukemia who fail to respond to imatinib. Like imatinib, dasatinib blocked MDSC expansion [41, 49] and could trigger the development of a broad repertoire of tumor-associated CD8+ tumor-infiltrating lymphocytes when associated with a DC-based vaccine in a melanoma model [54]. However, in several studies dasatinib also inhibited CD4+ and CD8+ T cell activation and proliferation in a dose-dependent manner [55]. The beneficial use of imatinib and dasatinib against MDSCs is unambiguous, but the consequences of their use on the T cell compartment remain unclear.
Many other TKIs have been shown to deeply affect MDSCs, their proliferation, differentiation, as well as their suppressive functions. Vemurafenib was approved for the treatment of unresectable or metastatic melanoma with the BRAF V600-activating mutation by the FDA in 2011. Vemurafenib could decrease the proportion and absolute number of M- and PMN-MDSCs as well as Tregs in melanoma both in mice models and human. Following a vemurafenib treatment, an increase in tumor-infiltrating CD8 T cells was observed and was correlated with a reduction in tumor size [56–58]. Approved by the FDA in 2013 to use against some B cell lymphoma, the Bruton tyrosine kinase (BTK) inhibitor ibrutinib could reduce MDSC accumulation in the tumor bed and reduce the expression of IDO. These effects are likely to be a direct consequence of the inhibition of BTK in MDSCs [59, 60].
Recently a growing interest regarding the effects of the PI3K/AKT/mTOR pathway on MDSCs has arisen. The mTOR pathway activation in both tumor cells and MDSCs seems favorable to MDSCs. Indeed, rapamycin, an inhibitor of mTOR, has been shown to significantly decrease MDSC number as well as the immunosuppressive functions of M-MDSCs in tumor-bearing mice. mTOR appears to be an intrinsic factor involved in the differentiation and suppressive functions of MDSCs [61, 62]. Moreover, activation of the mTOR pathway in cancer cells could also favor the recruitment and accumulation of MDSCs in a G-CSF-dependent fashion in human breast cancer [63]. However, we have to keep in mind that mTOR activation is also essential for T cell activation and mTOR inhibitor could have some deleterious effect on CD8 antitumor immune response. So far only rapamycin derivatives are used in clinic to block the PI3K/AKT/mTOR pathway, but other inhibitors targeting this pathway are in development. Such drugs should also be tested to address their capacity to inhibit MDSCs or reduce their number.
11.5 Other FDA-Approved Molecules with Impact on MDSCs
Molecules from different categories approved by the FDA are found to display activity against MDSCs. One of them is a phosphodiesterase 5 inhibitor named tadalafil. Tadalafil inhibited MDSC immunosuppressive functions via downregulation of iNOS and Arg-1, two key immunosuppressive enzymes of MDSCs [64]. In head and neck squamous cell carcinoma, tadalafil could reduce MDSCs and Tregs in both blood and tumor bed while increasing the concentration of CD8 T cells specific for tumor antigens in the blood [65, 66]. The dose at which tadalafil is used seems of importance as an important dose triggered off-target effects on PDE11 which may affect antitumor immunity by different ways [65]. A case report on a man with end-stage multiple myeloma showed that tadalafil reduced MDSC functions (Arg-1 and iNOS expressions downregulated) and established a durable anti-myeloma immune and clinical response although not complete [67].
CTL antigen-4 (CTLA-4) is a negative immune checkpoint expressed by T cells. Ipilimumab is a human antibody directed against CTLA-4, and it has been shown that in patients with metastatic melanoma, the frequency of PMN-MDSCs significantly decreased 3 weeks after a first treatment with ipilimumab [68]. In contrast, no impact on M-MDSCs was observed. Another study on melanoma patients showed a decrease in circulating MDSCs after an ipilimumab treatment and a positive association between decreased in MDSCs and a better PFS [69]. Complementary studies are required to understand the mechanisms by which ipilimumab affects MDSCs.
ATRA, for all-trans retinoic acid, is used to treat promyelocytic acute myeloid leukemia. This drug is also capable of inducing the maturation of MDSCs into DCs, macrophages, and granulocytes. As expected, the decrease in MDSCs by ATRA treatment improved CD4 and CD8 T lymphocyte tumor-specific response first in two mouse tumor models, DA3-HA adenocarcinoma and C3 fibrosarcoma [70], and then in patients with RCC [71] and small cell lung cancer [72].
11.6 Drugs in Developments
A broad spectrum of molecules from various origins have displayed an activity against MDSCs; they can either block the immunosuppressive functions of MDSCs, inducing their differentiation in dendritic cells or in M1-like macrophages, or deplete them.
In the MC38 colon carcinoma, the Lewis lung carcinoma and the EL-4 thymoma mouse models and in patients with RCC or soft tissue sarcoma, the triterpenoid CDDO-Me did not affect the size of the MDSC population but abrogated their immunosuppressive activities and improved immune responses [73]. Nitroaspirin also reduces MDSC functions by inhibiting NOS and Arg-1 activity, resulting in increased number and functions of tumor antigen-specific T lymphocytes [74]. The inhibitor of the ubiquitin receptor RPN13/ADRM1 RA190 was recently shown to lower the level of Arg-1, iNOS, and IL-10 in MDSCs. MDSCs treated by RA190 lost their capacity to suppress CD8+ T cells, thus enhancing antitumor immune response, in an ovarian mouse model [75]. TLR9 activation of MDSCs by CpG treatment was shown to block their suppressive activity on T cells in two mouse models of cancer and to induce MDSC differentiation [76]. The impact of CpG on MDSCs was confirmed in the Renca renal mouse tumor model [77]. On the opposite side, it is interesting to note that CpG emulsified in incomplete Freund’s adjuvant treatment expanded MDSCs and increased their expression of Arg-1 in aged mice free of tumor, suggesting contrasting effect of CPG on MDSCs [78].
Curcumin can differentiate MDSCs. Curcumin suppresses PMN-MDSC function and favors M-MDSC differentiation toward an M1-like phenotype [79] in a clusterin-dependent fashion [19]. Other molecules induce a depletion of MDSCs as the use of an antibody-targeting DR5, a death receptor present at the surface of MDSCs, can effectively eliminate them without affecting other myeloid populations and resulted in an enhanced antitumor immune response [80]. Beta-glucan-like curdlan can promote the differentiation of M-MDSCs into a mature CD11c+ F4/80+ population. That differentiation happens via a NF-κB-dependent dectin-1 pathway. A beta-glucan treatment diminished MDSCs in the tumor bed and increased infiltrated DCs and macrophages, leading to an enhanced CD8 and CD4 T cell responses and delayed tumor growth [81]. Two cationic polymers, cationic dextran (C-dextran) and polyethyleneimine (PEI), can differentiate MDSCs into an M1-like phenotype, decreasing IL-10 and TGF-beta production as well as suppressing Arg-1 expression while increasing M1-type cytokines production. This decrease in MDSC restored antitumor immunity and slowed tumor growth in the 4T1 mouse mammary carcinoma [82].
Depletion of MDSC can be achieved, thanks to a various set of molecule ever-expanding. However, the mechanisms behind that depletion are not always yet defined. A new therapeutic peptide, developed after identification and characterization of MDSC-binding peptides, depleted both monocytic and PMN-MDSCs in the blood and spleen of EL4 or EG7 thymoma-bearing mice and successfully delayed tumor growth [83]. The S100 protein family is a candidate target for this peptide, but a more thorough study may be needed in order to fully understand the mechanism of action of this peptide (Fig. 11.3).
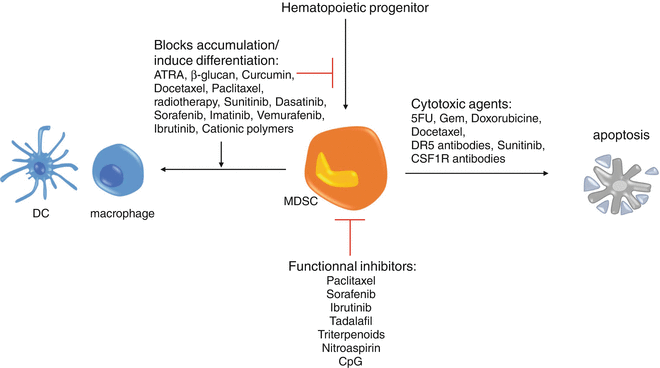
Fig. 11.3
Therapeutic approaches targeting MDSCs. There are three different ways to aim at MDSC. Molecules targeting MDSCs can directly kill them, like 5-FU or DR5 antibodies, or inhibit MDSC immunosuppressive functions as tadalafil does or also block MDSC accumulation or induce their differentiation like ATRA or curcumin does
CSF-1, also known as M-CSF, is overexpressed in many tumors and is a growth factor for M-MDSCs and macrophages. Several CSF-1 receptor inhibitors have been developed and, when tested in tumor-bearing mice, displayed the potency to deplete M-MDSC in tumor bed and spleen. Blockade of CSF1R increases antigen-specific T cell activity at the tumor site, delaying tumor growth in B16 melanoma [84, 85], RM-1, RM-9, and Myc-CaP prostate cancer-bearing mice [86].
Contradictory data can be found about histone deacetylase (HDAC) on MDSCs. It was first published that HDAC inhibition by TSA, a naturally occurring antifungal metabolite that potently inhibits HDAC, enhanced the expansion of MDSCs in a GM-CSF-dependent manner [87]. This was confirmed by another study showing that HDAC11 is a negative regulator of MDSC expansion and function and that EL4 tumor-bearing HDAC11 KO mice possessed a more suppressive MDSC population compared to wild-type mice [88]. However, in 2016, a team demonstrated that HDAC inhibitors depleted MDSCs induced by 4T1 mammary tumor both in vitro and in vivo in the spleen, blood, and tumor bed and increased the population of CD8 T lymphocytes. Interestingly, HDAC inhibitors also increased the apoptosis of MDSC precursor in the bone marrow, GR1+ cells [89]. Our understanding of the role of HDAC on MDSCs remains to be completed. Here the difference in models and inhibitors might be responsible for the difference in results, proving the complex interplay between MDSCs and the immune system state in tumor-bearing individuals.
11.7 Combination with Checkpoint Inhibitors
CTLA-4 and PD-1 (programmed death 1) are negative immune checkpoints regulating lymphocyte functions. CTLA-4 can be found in the cytoplasm of naïve T cells and is exported to the membrane after activation. Quantities of CTLA-4 found at the surface of activated T cells increase with their activation, allowing the creation of a negative feedback loop to avoid an overactivation of lymphocytes. CTLA-4 is a CD28 homologue binding CD80 and CD86 with a greater affinity than CD28. The ratio of CTLA-4/CD28 bound to costimulatory molecules will determine if the lymphocyte is activated or inhibited. Tregs express constitutively CTLA-4 which might play a part in the Tregs immunosuppressive functions. Blocking CTLA-4 results in an enhanced immune response explaining the interest it receives in oncology. PD-1 is found on highly activated T cells, NK cells, B cells, and monocytes. The binding of PD-1, with its ligands PD-L1 or PD-L2, found at the surface of tumor cells, and various tumor-infiltrated immune cells like MDSC and dendritic cells reduces T cell functions to avoid excessive immune responses [90]. PD-L1 plays a major role in the immunosuppression established in a tumor environment because of its expression on tumor cells and MDSCs; this is why the interaction PD-1/PD-L1 has led to the development of several antibodies aiming to block this interaction to restore potent antitumor immune responses [91]. Removing MDSC-dependent immunosuppression along with suppressing immune checkpoint blockade should induce a massive T cell response in tumor-bearing hosts. This is why several preclinical studies have tried to associate MDSC depletion or differentiation with antibody directed against negative checkpoint inhibitors (Fig. 11.4).
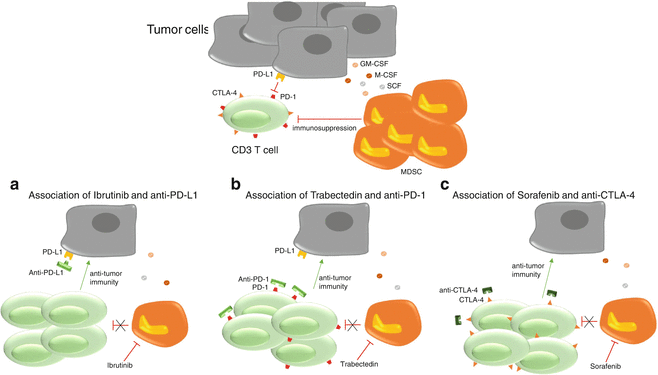
Fig. 11.4
Combination with checkpoint inhibitors. Tumor-induced immunosuppression is dependent on three aspects. The direct immunosuppression mediated by tumor cells expressing PD-L1 on PD-1+ T cells, the induction of MDSCs by tumor cells and MDSC-dependent immunosuppression. By associating a molecule depleting MDSCs with a checkpoint inhibitor, it is possible to restore a potent antitumor immunity. (a) Ibrutinib depletes MDSCs, while the anti-PD-L1 blocks the PD-1/PD-L1 immunosuppression. This allows T cells to proliferate and establish an antitumor immunity, preventing tumor growth. (b) Trabectedin can directly suppress MDSCs and, when associated with an anti-PD-1, restores a proper T cell-mediated antitumor immunity. (c) The action of sorafenib against MDSCs in association with an anti-CTLA-4 allows T cells to develop an antitumor immune response leading to a reduced tumor growth
Ibrutinib, an inhibitor of BTK and ITK (interleukin-2-inducible T cell kinase) known to reduce MDSC accumulation in the tumor bed [59, 60], can enhance therapeutic antitumor immunity when associated with a PD-L1 treatment in the A20 lymphoma but also the CT26 colon carcinoma and 4T1 breast carcinoma models [59]. This was later confirmed by another study where the association of an anti-PD-L1 with ibrutinib almost completely abrogated tumor growth of 4T1 breast cancer [60]. In a model of ovarian cancer, trabectedin was associated with an anti-PD-1. This association cured about half of the mice and induced a strong tumor-specific immunity from CD4 and CD8 T cells. CD8 T cells exhibited tumor antigen-specific responses, and an increase in IFN-gamma was observed along with a reduction in immunosuppressive populations. Interestingly, in vivo trabectedin might be responsible for a rise in PD-L1 expression within tumor explaining the improved efficacy of the association over single therapies [92].
High-dose ionizing irradiation (IR) results in direct tumor death and is used in many cancers. In the TUBO breast cancer and MC38 colon cancer models, IR also decreased the population of MDSCs but increased PD-L1 expression inside tumors. To overcome this issue, IR was used along with an anti-PD-L1. This association reduced MDSC population to close to zero percent in the tumor bed while enhancing cytotoxic CD8 T cells in a synergistic manner, delaying tumor growth [93]. In HPV-related oropharyngeal cancer, radiotherapy is often being associated with chemotherapy to treat patients. In a clinical trial, authors observed HPV-specific T cell responses in 13/18 patients prior to treatment. This immune response was lost in 10/13 patients within 3 months after chemoradiotherapy (CRT). CRT decreased circulating T cells and increased the MDSC population. PD-1 expression on CD4 T cells was also enhanced after CRT. The use of a PD-1 blocking antibody in ex vivo culture restored the HPV CD4 T cell-specific response, further encouraging the study of such association to help improve patient treatments [94].
As previously seen, sorafenib can effectively deplete MDSCs. It was associated with an anti-CTLA-4 in a RENCA mouse model and whereas the monotherapies did reduce tumor growth, the combination displayed a synergistic effect with the highest rate of tumor rejection and a strong increase in infiltrating CD4 and CD8 T lymphocytes in the tumor bed [95]. Unfortunately, the number of MDSCs was not assessed. There is an ongoing phase I study about the combination of sorafenib and ipilimumab (anti-CTLA-4) in patients with advanced hepatocellular cancer with a stable disease that should give us more information.
Ipilimumab is also often associated with an anti-PD-1 named nivolumab, and this combination is now used as standard treatment of metastatic melanoma in patients. While ipilimumab could decrease MDSC number, no study thoroughly examined the impact of this association on MDSCs. However, an increase of CD4 and CD8 T lymphocytes to MDSC ratio was observed in the mouse melanoma model B16 using a combination of anti-PD-1 and anti-CTLA-4 [96].
Related posts:
 Peptide-Based Therapeutic Cancer Vaccines
Peptide-Based Therapeutic Cancer Vaccines
 The Human Tumor Microenvironment
The Human Tumor Microenvironment
 PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
 Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
 Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
 Immune Monitoring of Blood and Tumor Microenvironment
Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


