Cushing’s syndrome is a symptomatic manifestation of a wide variety of disease processes culminating in hypercortisolism, only a few of which, including ectopic ACTH syndrome (EAS), necessitate the attention of a surgical oncologist.1 In the classical setting of EAS, the diagnosis begins under the supervision of an endocrinologist, and the careful application of a diagnostic algorithm of laboratory testing, imaging, and/or interventional procedures often helps identify a source.2 However, EAS can also present in a paraneoplastic fashion in a patient with a known malignancy, and surgical oncologists may be called upon to identify a potential source. This chapter will briefly review the physiology of the hypothalamic-pituitary-adrenal (HPA) axis and Cushing’s syndrome, discuss the diagnosis of EAS and its descriptive classifiers, and outline management strategies for the care of patients with EAS.
The HPA axis is complex feedback regulatory mechanism by which the body maintains cortisol homeostasis and responds to episodes of stress. Neuroendocrine cells within the paraventricular nucleus of the hypothalamus secrete corticotropin-releasing hormone (CRH) and vasopressin. These peptides stimulate the anterior lobe of the pituitary gland to release adrenocorticotropic hormone, which in turn, stimulates the adrenal cortex to produce glucocorticoid hormones, including cortisol. The presence of cortisol downregulates production of CRH and ACTH in the hypothalamus and pituitary gland, respectively, completing a negative feedback loop with a somewhat predictable circadian rhythm of circulating serum cortisol in the absence of dysregulation of the HPA axis (Fig. 51-1).3
FIGURE 51-1
The hypothalamic-pituitary-adrenal axis. A. In a physiologic state, release of cortisol from the adrenal cortex provides negative feedback to the hypothalamus and pituitary (green arrows), to decrease the release of corticotropin-releasing hormone (CRH) and adrenocorticotropic hormone (ACTH), respectively. B. In EAS, cortisol feedback inhibits release of CRH and pituitary ACTH, but the ectopic source of ACTH is unaffected, resulting in adrenal hyperplasia and large excess circulating cortisol. Ectopic source depicted by red mass and superphysiologic amounts are depicted by the red arrows.
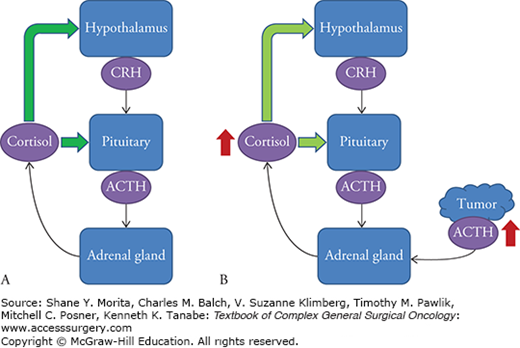
Dysregulation can occur as a normal physiologic response to stress, as a result of exogenous administration of glucocorticoids, or by overproduction of CRH or ACTH by tumors. When sustained dysregulation leads to hypercortisolism, specific metabolic effects can be seen, identified as Cushing’s syndrome. The most common cause is exogenous glucocorticoid usage exceeding physiologic requirements and can often be ascertained by obtaining a careful medication history. Endogenous forms of the syndrome are divided into ACTH-dependent and ACTH-independent causes. Overall, ACTH-dependent cases account for 80% to 85% of endogenous causes, the majority of which are Cushing’s disease; ectopic ACTH production is responsible for about 20% of ACTH-dependent Cushing’s syndrome.1
The first case of EAS was described in 1928, though it was initially noted as an association of two independent conditions.4 As more cases appeared in the literature, there was a strengthening of the correlation between various cancers and hypercortisolism, and an astute endocrinologist published his findings on this new syndrome of malignant ACTH secretion in 1962.5 In the ensuing years, clinicians have refined and classified the syndrome, but his initial observations remain true: certain tumors acquire the ability to secrete ACTH, circulating levels of ACTH are sufficient for adrenal hyperplasia and hypercortisolism inducing features of Cushing’s syndrome and suppressing pituitary synthesis and secretion of ACTH, and this process is not suppressed by high levels of synthetic steroid.6 EAS has now been described in an ever-widening variety of tumors with a clinical heterogeneity seemingly dependent on the biological aggressiveness of the underlying tumor.
While many of the intracellular mechanisms remain under investigation, regulation of the ACTH-precursor, proopiomelanocortin (POMC), may lie at the center of the EAS. The POMC gene lies on the long arm of chromosome 2 and consists of three exons and two large introns.7 At least three promoter regions have been identified, each generating mRNA of differing lengths. The classical pituitary promoter has been most extensively studied, and its eventual peptide product is then cleaved into a number of active hormones, including ACTH. Transcription of an upstream promoter has been identified in cell lines derived from patients with EAS, while a downstream promoter has been identified in normal nonpituitary tissue, suggesting low levels of POMC gene expression in a wide variety of tissue. While this may provide a basis for the heterogeneity of the neoplasms responsible for EAS, their oncogenesis is still unclear.8
The post-translational processing of nonclassical peptide products may differ, overwhelming intracellular machinery and leading to high circulating levels of precursor molecules (pro-POMC and POMC).9 Clinically, significant differences have been noted in precursor:ACTH ratios in patients with EAS compared to Cushing’s disease. Precursor molecules are much less potent than ACTH in stimulating the adrenal cortex to produce cortisol; however, minimal activity combined with high circulating levels may be the etiology for EAS in patients with low normal ACTH levels.10 Even more rare, and less well characterized, is a secondary elevation of ACTH from a CRH-producing tumor, often diagnosed via immunohistochemistry and clinically indistinguishable from EAS.11
The diagnosis of ectopic ACTH secretion begins with the identification of signs and symptoms of Cushing’s syndrome, though the EAS patient subpopulation tends to be older and more evenly gender-distributed (Table 51-1).12 Biochemical confirmation of endogenous hypercortisolism is obtained through measurement of elevated urinary-free cortisol, lack of cortisol suppression after low-dose dexamethasone administration, elevated late-night salivary cortisol, or elevated midnight serum cortisol. Plasma levels of ACTH can then be obtained to determine an ACTH-dependent or ACTH-independent etiology. A normal to elevated ACTH level indicates an ACTH-dependent cause, and the source must be then determined to be pituitary (Cushing’s disease) versus nonpituitary (EAS). Noninvasive diagnostic tests, including a CRH-stimulation and a high-dose dexamethasone suppression test, are usually performed in conjunction with a pituitary MRI. If no obvious pituitary lesion is identified, bilateral inferior petrosal sinus sampling is considered the gold standard for ruling out a pituitary source. The details of this complete diagnostic algorithm are beyond the scope of this chapter1; however, when Cushing’s disease has been ruled out, the tentative diagnosis is EAS and a search for a tumor source begins. The majority of tumors responsible for EAS will be found in the neck and thorax,2,12–14 though case reports of unusual tumors continue to appear in the literature (Table 51-2).4,15–35
Clinical Features of EAS Present in Majority of Patientsa
| Weakness due to proximal myopathy |
| Hypertension |
| Menstrual irregularity |
| Hirsutism |
| Osteopenia or osteoporosis |
| Hypokalemia |
| Weight gain |
| Psychiatric disorders |
| Bruising |
| Infections |
| Bronchial carcinoid | 21–38.9% |
| Small cell lung cancer | 3.3–21% |
| Thymic carcinoid | 4.7–6.9% |
| Medullary thyroid cancer | 2.2–11.6% |
| GI or Pancreatic neuroendocrine tumor (NET) | 8.9–17% |
| Pheochromocytoma/paraganglioma | 0–5.6% |
| NET of unknown primary | 5–14.4% |
| Occult | 9.3–18.9% |
| Othera | 2.2–9.3% |
Though earlier published studies discuss possible localization of tumors with chest radiograph alone, today patients are likely to proceed directly to axial imaging. Thin-cut, multislice computed tomography (CT) of the chest, abdomen, and pelvis with contrast enhancement is the preferred initial diagnostic imaging, with magnetic resonance imaging (MRI) reserved for equivocal cases. Of the 40 patients described retrospectively by Isidori et al,14 CT was able to identify a tumor in 23 of the 28 patients imaged. In a larger series by Ilias et al,12 the combination of CT and MRI was able to identify tumor in 67 of 73 patients. However, both modalities yielded false-positive results, and many patients underwent additional whole body functional imaging.
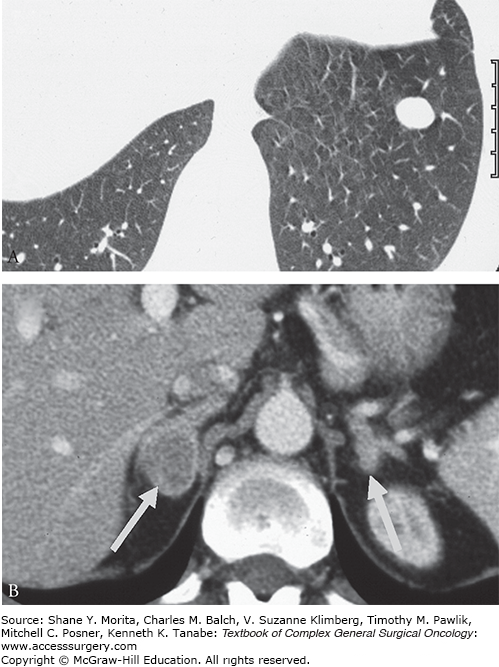
Bronchial carcinoids pose a diagnostic dilemma as these functional tumors can be quite small in size. In a review by Sookur et al,37 there were no distinguishing CT features between functioning and nonsecretory tumors, other than a differential in size, with functioning tumors ranging from 4 to 10 mm and larger nonsecretory masses ranging from 12 to 40 mm, a finding presumed to be biased by earlier presentation of functioning nodules. Tumors were peripherally located, well-defined, ovoid enhancing masses without evidence of calcification (Fig. 51-2A).38 In challenging cases, prone imaging helped identify small lesions in dependent portions of the lung. MRI can be used to clarify equivocal findings in the pulmonary hilum as bronchial carcinoids have high T2 and STIR signal, in contrast to pulmonary vasculature.
FIGURE 51-2
Axial imaging of a bronchial carcinoid causing EAS. A. A well-defined, ovoid nodule in the periphery of the left lung. B. Secretion of ACTH can lead to bilateral nodular adrenal hyperplasia. (Reproduced with permission from Rockall AG, Reznek RH. Imaging of neuroendocrine tumours (CT/MR/US), Best Pract Res Clin Endocrinol Metab. March 2007;21(1):43–68.)
Thymic carcinoids account for a small percentage of EAS and are seen as soft-tissue masses in the anterior mediastinum. Those associated with EAS may present with proximal lymph node involvement and may show contrast enhancement and calcification. In younger patients, functional imaging can help differentiate these tumors from normal remnant thymic tissue.37
Pancreatic NET can also cause EAS, however this type tends to be more advanced on presentation, often with metastatic liver deposits. Nonetheless, pancreas-specific triple phase thin-slice CT protocols can aid the delineation of tumor from normal tissue. These scans should include unenhanced images, arterial phase images acquired 30 seconds after intravenous contrast, and portal venous phases 60 to 70 seconds after contrast. Tumors have been seen throughout the pancreatic parenchyma and tend to be hypervascular in nature, appearing best on arterial phase images.37
A hallmark of EAS may be the combination of finding a suspicious tumor with marked hyperplasia of the adrenal glands (Fig. 51-2B).38 This finding is present in more patients with EAS and to a greater degree than in those patients with Cushing’s disease (90% vs. 63%). There is wide variation from diffuse smooth hyperplasia to nodular contouring, with 19% demonstrating a nodule ≥1 cm.37
While often used to corroborate equivocal cross-sectional findings, functional imaging can also be used in patients with persistently negative findings. Pacak et al39 performed a prospective trial to evaluate 18-fluorodeoxyglucose (FDG) positron emission tomography (PET) and a somatostatin analog PET at low and high doses in comparison with axial imaging. In a small sample size (n = 17), the conclusion was that functional imaging with a somatostatin analog at low doses could be considered part of an initial evaluation, as it had the same sensitivity, 53%, as CT. High dose should only be considered after a negative low-dose scan. In that study and other case reports, traditional FDG PET was not helpful in finding occult lesions, presumed to be a function of the relative low proliferative rate of carcinoid lesions (Fig. 51-3).40
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


