1. Disease status
(a) How was the diagnosis made?
(b) Consideration of variant forms of diabetes (e.g. pancreatic insufficiency secondary to pancreatic disease, MODY, etc.)
(c) Duration of disease
i. Pancreatic functional reserve
ii. Serum C-peptide level
(d) Complications
i. Retinopathy
ii. Nephropathy
iii. Neuropathy
iv. Macrovascular disease
(e) Current control – HbA1c
(f) History of hypoglycaemia
2. Comorbidities
(a) Metabolic syndrome
i. Hypertension
ii. Dyslipidaemia
(b) Fatty liver disease
(c) Depression
(d) Degenerative joint disease
(e) Hypothyroidism
(f) Sleep apnoea
3. Current diabetes therapy
(a) Diet and exercise controlled
(b) Monotherapy
(c) Insulin therapy
4. Concomitant medications
(a) Antihypertensive agents
i. Diuretics
ii. β-adrenergic blockers
iii. ACE inhibitors
iv. ARBs
(b) Treatments for dyslipidemia
i. Statins
ii. Fibrates
iii. Fish oil
(c) Aspirin
(d) Pain medication (NSAIDs, opiates)
(e) Antidepressants
i. SSRIs
(f) Hormonal therapy
i. Thyroid hormone replacement
ii. Oestrogen/progesterone replacement
iii. Oral/implantable contraceptives
iv. Testosterone replacement
(g) Nutraceuticals/vitamins/herbal treatments
Assessment of disease status is the first criterion. For most patients, differentiating between type 1 and type 2 diabetes is straightforward. A well-performed medical history, conducted by qualified medical personnel ad accompanied by supporting medical evidence (use of glucose-lowering medication, laboratory findings consistent with the diagnosis of diabetes mellitus, or medical records), is usually sufficient. While medical records can be very helpful, obtaining such records in a timely fashion can be challenging in early phase clinical trials. Additionally, medical records not infrequently contain information that is discrepant to that obtained from the patient; the judgement of the investigator is necessary to determine whether or not a patient is eligible for participation in a particular study. Occasionally, the diagnosis of diabetes is in question – particularly for individuals who have achieved excellent metabolic control through non-pharmacological methods, i.e. diet and exercise, and no longer appear to meet diagnostic criteria for diabetes. The success of gastric bypass surgery on remission of diabetes is relevant, although patients with a history of bariatric surgery would be excluded from most early phase diabetes drug studies because of the potential impact of the surgery on absorption of the investigational product and the known impact of these procedures of the gastrointestinal hormonal milieu and glucose metabolism [27]. Additionally, the category of diabetes is sometimes in question, e.g. in individuals with type 1 diabetes mellitus who may be initially misdiagnosed as having type 2 diabetes, particularly if they are older at the time of diagnosis [28]. Alternatively, individuals with maturity-onset diabetes of the young (MODY) may be misdiagnosed as having type 1 diabetes [29]. Measurements of islet autoantibodies, both glutamate decarboxylase (GAD) and islet antigen-2 (IA-2) autoantibodies, highly prevalent in type 1 diabetes and not usually present in MODY, can be helpful in differentiating these forms of diabetes [30, 31]. Additionally, the presence of GAD antibodies in individuals with previously diagnosed type 2 diabetes mellitus (perhaps as high as 10–25 %) who did not require insulin therapy in the first 6 months after diagnosis suggests that the correct diagnosis is latent autoimmune diabetes in adults (LADA) [32]. Including these assessments can be useful if the differentiation of the subtypes of diabetes is critical. Additionally, the measurement of serum C-peptide can be of value in helping to support the diagnosis of type 1 diabetes mellitus or the lack of residual β-cell function [33]. Low fasting serum C-peptide levels (<0.3 nmol/L) are indicative of absolute insulin deficiency and type 1 diabetes mellitus. The use of serum C-peptide measurement in studies involving patients with type 2 diabetes is more useful in determining residual β-cell function than using duration of diagnosis, as the date of diagnosis poorly reflects duration of disease [34].
Included in the assessment of disease status is an assessment of the presence or absence of long-term vascular and neurological complications of diabetes [35]. The presence of advanced complications, particularly proliferative retinopathy; diabetic nephropathy with significant proteinuria or renal insufficiency; diabetic neuropathy, particularly with impaired gastric emptying; or clinically significant macrovascular disease are likely to be exclusionary conditions for individuals with diabetes wishing to participate in early phase clinical trials. The lack of experience with patient exposures to the new investigational product may place these more vulnerable patients at increased levels of risk. Impairment of drug absorption, metabolism, or excretion because of underlying complications of diabetes could have substantial impact on initial assessments of drug pharmacokinetics, from which many early decisions in drug development are made. Additionally, the occurrence of a serious or severe adverse event, which may or may not be related to the compound under investigation, in a subject participating in an early phase trial can significantly delay or even curtail drug development.
Glycaemic Control
Essentially every volunteer participating in an early phase clinical trial involved with carbohydrate metabolism has an assessment of his/her glycated haemoglobin (HbA1c) level. Figure 9.1 demonstrates the ranges of HbA1c for our volunteer database; only about one quarter of volunteers with type 1 diabetes have an HbA1c of <7.0 % (<53 mmol/mol) and only about one third of volunteers with type 2 diabetes are similarly well controlled. Most clinical trials have defined upper and lower boundaries for inclusion in studies. These cutoffs serve several purposes: safety, uniformity of effects on outcome measures, and efficacy assessments. Most safety considerations are concerned with putting trial participants at risk for hypoglycaemia if they enter studies with a normal or only modestly elevated HbA1c. Lower cutoffs are usually at 6.5 or 7 % (48–53 mmol/mol) whereas upper boundaries are generally in the 10–11 % (86–97 mmol/mol) range. Individuals who are above these levels are excluded for ethical reasons – these patients should receive an urgent referral for appropriate healthcare. Such patients face risks for worsening hyperglycaemia should the compound under study be ineffective or the subject is randomized to a placebo treatment arm. This risk can be accentuated for proof-of-concept studies where the participation period of any given subject could extend to 3 months or more. Additionally, concerns of detrimental effects on islet β cells from hyperglycaemia (so-called glucotoxicity) [36] and variable treatment effects because of variability in the level of metabolic control contribute to these limits. While the primary purpose of early phase studies, particularly first-in-human, single ascending dose, and multiple ascending dose studies, is assessment of safety and tolerability, increasingly, sponsors look to these studies to provide early efficacy signals. In general, HbA1c is a poor indicator of efficacy in these early trials because of the treatment time needed to see improvements in HbA1c. For trials that extend over several weeks, an alternative measure of glycaemic control (1,5 anhydroglucitol, fructosamine) may be of value [37, 38].
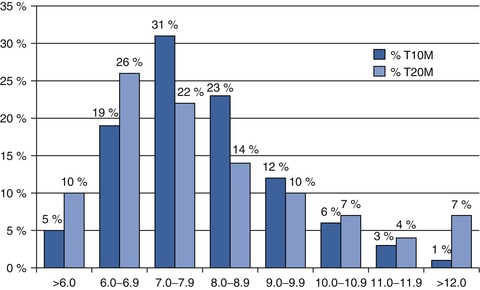
Fig. 9.1
HbA1c distribution derived from a subset of US database of early phase trial volunteers. Data represents a subset of patients with type 1 diabetes (n = 874, 53 % of total population) and a subset of patients with type 2 diabetes (n = 2,889, 33 % of total population) for whom clinic-measured HbA1c results were available
Risk of Hypoglycaemia
A related factor to diabetes control that must be assessed in the medical evaluation is the patient’s history regarding hypoglycaemia. It is well documented that the risk for hypoglycaemia often rises in concert with attempts to lower HbA1c. Individuals with tighter diabetes control and longer duration of disease tend to have more episodes of hypoglycaemia [39], and recurrent bouts of hypoglycaemia put patients at risk for so-called hypoglycaemia unawareness [40]. Most protocols appropriately exclude individuals with recent episodes of severe hypoglycaemia (defined as hypoglycaemia rendering the patient unable to treat himself/herself), and individuals with hypoglycaemic unawareness must be excluded from early clinical trials where the glucose-lowering capabilities of new compounds are not yet defined. Because investigators and study staff (as well as the participating subjects) are usually blinded to treatment randomization and potentially unblinding pharmacodynamic results, subjects must be able to alert staff promptly to symptoms of hypoglycaemia.
Cardiovascular Disease
Enrolling patients into clinical trials implies the willingness to consider the presence of comorbidities related to the disease state under investigation and the concomitant medications that might be required to treat either the primary disease or the comorbidities. Frequent comorbidities in individuals with diabetes are the known associated complications of diabetes as discussed above, many of which could exclude a patient from participating in an early phase trial by virtue of the unacceptable safety risk. Other comorbidities, particularly in patients with type 2 diabetes, include those related to the metabolic syndrome – obesity, hypertension, and dyslipidaemia (see below). Of particular importance in patients with any disorder of carbohydrate metabolism is atherosclerotic coronary heart disease. It is known that any abnormality in glucose tolerance increases the risk of coronary heart disease along a continuum [41], even without the presence of diabetes mellitus. As a minimum, individuals must be screened with a careful medical history and a 12-lead electrocardiograph (ECG). Any suggestion of previously undiagnosed disease (chest pain, both typical and atypical, shortness of breath, exercise intolerance, signs of congestive heart failure, or resting ECG abnormalities suggestive of ischaemia or prior myocardial infarction) requires further evaluation. For patients with a known history of coronary heart disease, relevant medical records must be obtained. While a history of a cardiac event does not invariably lead to exclusion from participation in all early phase trials, such individuals should be excluded from first-in-human studies until there is a greater understanding of the risk associated with the compound under development. Alterations in routine laboratory measurements (e.g. platelet counts, lipid profiles, clotting function), evidence of ECG conduction defects, and individual risk for hypoglycaemia need to be carefully considered on an individual basis. One of the potential benefits for individuals screening for trials is that previously undiagnosed comorbidities can be revealed and then the patient referred for additional evaluation and treatment.
Prolongation of the electrocardiographic QT/QTc interval is a standard exclusion criterion for early phase studies of new diabetes drugs. The need to ensure cardiovascular safety of novel glucose-lowering drugs, as is the case for all new non-antiarrhythmic drugs, starts with the ‘thorough QT/QTc’ study [42]. The ‘International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use’ (ICH) issued the E14 clinical guidance in 2005 which was quickly implemented in the USA and Europe [43]. The thorough QT/QTc study has become a standard component of the clinical development for new molecular entities. The link between QT prolongation and drug therapies was established following reports of deaths in patients treated with cisapride, a gastrointestinal prokinetic drug that was widely used in the treatment of diabetic gastroparesis [44, 45]. With some exceptions, such as cytotoxic cancer drugs, thorough QT/QTc studies are usually performed in healthy volunteers. The QT interval, which is measured from the beginning of the QRS complex to the end of the T wave, represents the duration of ventricular depolarization and subsequent repolarization. A delay in cardiac repolarization creates an electrophysiological environment that favours the development of cardiac arrhythmias, most clearly torsade de pointes [44]. QTc refers to the correction of QT interval for heart rate [42]. Cardiovascular toxicity is a major concern for many classes of drugs during preclinical and clinical development and may lead to post-approval withdrawal of medicines [46]. Since 2008, in the wake of the controversy concerning a reported increase in myocardial infarction with rosiglitazone [47, 48], the FDA has mandated rigorous demonstration of cardiovascular safety of new diabetes drugs [49, 50]. Before rosiglitazone, the cardiovascular safety of diabetes drugs had not been closely scrutinized [51]. While the FDA subsequently rescinded its restrictions on the use of rosiglitazone [51, 52], a major change in policy towards the assessment of cardiovascular risk of new diabetes drugs was already firmly in place. The European Medicines Agency (EMA) has also issued guidance on cardiovascular safety requirements of diabetes drugs [53]. New therapies directed towards glycaemic control may have cardiovascular benefits; however, a reduction in clinical events remains to be proven [54]. According to the 2008 FDA guidance, phase 2 clinical studies may be included in a meta-analysis of premarketing cardiovascular safety studies [55]. The cautious new regulatory environment has increased awareness of the need to de-risk new drugs for cardiometabolic disorders. This needs to happen before expensive late phase clinical studies are initiated [12]. Early phase clinical studies offer an opportunity to identify early signals of cardiovascular effects of new drugs for diabetes and obesity that may inform clinical development decisions. Differences in risk of diabetes-associated cardiovascular disease outcomes between men and women are recognized and remain incompletely understood [56, 57]. Recent data suggest heterogeneity in the effects of certain glucose-lowering drugs on cardiac metabolism and function between men and women [58]; this observation merits further study.
Fatty Liver Disease
While the presence of fatty liver within the obese and type 2 diabetes populations is well known, patients with type 1 diabetes may also have an increased prevalence of fatty liver [59, 60]. Elevations in transaminases are commonly seen in individuals with diabetes [61] and are seen frequently in those screening for participation in early phase clinical trials. Other causes of elevated liver function tests need to be excluded (e.g. alcohol, hepatitis, human immunodeficiency infection). Careful consideration is needed concerning the inclusion of subjects with modest elevations of transaminases in first-in-human trials. Since Hy’s law [62, 63] is commonly utilized to assess the risk of liver injury from a new compound in development, subjects should not be included in early phase studies with transaminases >3× upper limit of normal (ULN) as defined by the particular laboratory service provider; consideration should be given to setting the limit for inclusion to <1.5 or 2× ULN. Enrolling subjects with higher transaminases risks exceeding the limits included in Hy’s law related to variability that is commonly seen with transaminases and might not be attributable to the compound under investigation. One additional abnormality this is commonly seen is an elevation in indirect bilirubin levels in Gilbert’s syndrome, a variant of glucuronyl transferase that affects ~5–10 % of the total population. Most commonly, this is initially noted as an elevation in total bilirubin concentration since fractionation of bilirubin is frequently omitted in screening panels. Fasting or even modest dehydration can raise the level of indirect bilirubin in individuals with Gilbert’s syndrome. Fractionation of bilirubin can be helpful in excluding more serious disorders. Most early phase studies allow subjects with Gilbert’s syndrome with modest elevations in indirect bilirubin unless there are specific compound-related effects under consideration.
Diabetic Nephropathy
Diabetic nephropathy is the most common cause of chronic kidney disease in the USA [64]. Patients with either long-duration type 1 diabetes or type 2 diabetes may be affected. Depending on factors including age, duration of diabetes, and long-term glycaemic control, approximately 20–40 % of patients with type 2 diabetes may incur a moderate or severe deterioration of renal function. The clinical stages range from degrees of albuminuria to end-stage chronic kidney disease. Albuminuria not only is regarded as a marker and predictor of progressive renal disease but also denotes an increased risk of cardiovascular events [65]. A decline in glomerular filtration is not invariably paralleled by the emergence and increase of urinary albumin excretion; dissociations between albuminuria and decreased glomerular filtration rates (GFR) may be observed [66], Unless testing a new therapy that is focused on preventing, retarding, or reversing nephropathy, good renal function, i.e. estimated GFR (eGFR) calculated by the Modification of Diet in Renal Disease (MDRD) [67], is usually specified as an inclusion criterion for participation in early phase studies of new treatments for diabetes. Renal impairment necessitates dose reduction or avoidance of most classes of glucose-lowering agents to maintain a favourable risk-benefit equation, primarily with respect to the risk of hypoglycaemia [68]. Studies of PK in subgroups of patients with varying degrees of renal impairment will usually form part of the clinical development programme of a novel glucose-lowering drug [69].
Other Comorbidities
Additional clinically evident or subclinical comorbidities commonly encountered in diabetes populations screening for clinical trials are degenerative joint disease, hypothyroidism, sleep apnoea, and depression. Each of these, apart from the associated concomitant medications, has potentially important practical implications for participants in early phase trials. Patients with degenerative joint disease may have mobility issues that make completing study assessments more challenging; affected patients may need special accommodations when resident to manage their disabilities. Subjects with hypothyroidism need to be tested to be sure that their thyroid function tests are within an acceptable range and that their medication regimen is stable. Patients with sleep apnoea who require continuous positive airway pressure (CPAP) or other support will need to have this treatment provided while accommodated in a clinical trial unit, assuming that the therapy is compatible with the protocol. Depression is common among individuals with diabetes, with a meta-analysis revealing an overall prevalence of depression of ~25 %, twice that of the nondiabetic population [70]. This has at least two significant implications for early clinical trials: firstly, many of these patients will be using antidepressant medication, most commonly selective serotonin reuptake inhibitors (SSRIs), and secondly, changes in mood are of particular interest for compounds that could act centrally to control appetite, obesity, and even glucose counter-regulation [71]. The demise of the first selective endocannabinoid receptor antagonist, rimonabant, as a treatment for obesity resulted from adverse effects on mood and increased risk of suicidal ideation [72]. Patients with a past or current history of depression should be excluded from early trials where these issues might be anticipated; early trials should also be designed to include assessments for central effects of novel therapies.
Concomitant Medications
While it is not possible to cover all of the concomitant medications that patients with type 1 or type 2 diabetes might be taking at the time of screening, the majority of individuals volunteering for clinical trials will be taking some concomitant therapy, particularly those with type 2 diabetes. In our database of >20,000 active research subjects with maintained medical records, only 7 % of patients with type 2 diabetes take no medications whereas 64 % of patients with type 1 diabetes take no medication in addition to their insulin. As noted above, comorbidities are commonplace in these populations and medications are often recommended for prophylaxis of such comorbidities for all patients with diabetes, principally statins and other lipid-modifying agents, angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), and aspirin. Table 9.1 contains the classes of medications that are commonly encountered. These should be considered for every protocol written for early phase metabolic trials including patient populations as well as healthy volunteers. Exclusion of these medications significantly affects the ability of clinical sites to enrol subjects in trials; additionally, these medications will be used by the target population, and issues with drug-drug interactions should be identified as early as possible. However, drugs known to be strong inducers or inhibitors or substrates for metabolic pathways utilized by the compound in development are usually excluded from early phase trials. The FDA has provided guidance for these issues, along with tables of substrates, inhibitors, and inducers [73, 74]. A few medications deserve special mention: oral or injectable corticosteroids are usually excluded if patients have had recent exposure (usually within 3 months of study drug dosing) because of the well-recognized adverse effects on insulin sensitivity and carbohydrate metabolism [75]. Inhaled or topical corticosteroids can usually be allowed because the very low circulating levels that are achieved by these routes are not considered to have any significant impact on glucose tolerance. β-adrenergic blockers are also frequently excluded because of concerns, focused on non-selective agents, that include impairment of glucose and lipid metabolism and masking symptoms of iatrogenic hypoglycaemia [76, 77]. Acetaminophen is generally allowed as needed up to certain doses (usually 1,000–1,500 mg/day) to manage headache or other common discomforts that can be present during participation in a clinical trial, as are routine vitamin therapies at therapeutic doses (e.g. once-a-day multiple vitamin preparations). Most other vitamin and herbal preparations can and probably should be discontinued after screening until study end.
Type 1 Diabetes
Patients with type 1 diabetes mellitus commonly participate in very early development of new insulins and new devices that are developed for assessment and treatment of diabetes (insulin pumps, continuous glucose monitors, and blood glucose meters). The lack of, or very low levels of, endogenous insulin makes the interpretation of studies much simpler, as PK assessments reflect only exogenously administered insulin, and there is no risk of stimulation of endogenous insulin secretion during glucose clamp studies, for example. While the type 1 diabetes study population tends to skew towards the younger age range, the risk for hypoglycaemia and hyperglycaemia is much greater in this patient population; clinical trials must be carefully designed to take due account of these risks. When a new insulin is being evaluated, care must be taken to avoid any interference from routine insulin therapy. Long-acting basal insulin preparations must be discontinued 48 or more hours (depending on the half-life of the basal insulin currently being used by the patient) prior to study dosing. This can be accomplished by initially switching the patient from a basal/bolus regimen to one using isophane (NPH, neutral protamine Hagedorn) insulin and a rapid-acting insulin analogue and then admitting the patient to the clinical unit at least 24 h prior to planned study drug dosing. The last dose of NPH should be administered subcutaneously no later than 24 h prior to study drug dosing, and the last dose of rapid-acting insulin should be administered no later than 6 h prior to dosing. Patients with type 1 diabetes using continuous subcutaneous insulin infusion pumps should discontinue their pumps at this time. Whenever insulin therapy is altered, careful supervision, close monitoring, and ready access to expert advice must be in place to avoid metabolic decompensation. Competence in managing insulin therapy is a prerequisite for investigators supervising clinical trials of diabetic patients.
Two other critical factors in designing studies with patients with type 1 diabetes are ensuring comparability of baseline measurements of glucose and a stable metabolic milieu. Both of these can be accomplished by admission of patients to the study unit at least 24 h prior to important study procedures, which allows for tight control of meals and blood glucose. By giving the last dose of NPH insulin 24 h prior to study drug dosing, administering rapid-acting insulin prandially and as needed until ~12 h prior to study drug dosing, and then instituting an overnight intravenous insulin infusion along with the fasting state, good control can usually be accomplished with modest amounts of insulin. The intravenous insulin infusion can be discontinued at or prior to study drug dosing or with the apparent onset of study drug action or even maintained at a stable infusion rate, depending upon the study design and compound under investigation. Care should be taken to avoid discontinuation of intravenous insulin prior to the onset of study drug action since it may result in hyperglycaemia with unstable baseline conditions.
Maintenance of diabetes control in an early phase clinical trial unit can be a complex undertaking, depending on the compound under study and the particulars of the trial protocol. Use of standardized diets, with known nutrient content, can simplify prandial insulin dosing, particularly for those patients who utilize insulin-to-carbohydrate ratios to determine their mealtime doses. Input from the patients themselves regarding appropriate dosing for meal coverage will usually be helpful for the investigators; this information should be sought unless the protocol dictates the dosing. Upon release from the clinical unit, study volunteers are usually returned to their usual insulin regimen. Frequent monitoring should be performed and adjustments to insulin dosing should be made as needed to accommodate any lingering drug and/or diet effects. Follow-up of the patients should continue until their metabolic control is back to their individual baseline and can usually be accomplished by telephone contact; avoidance of hypoglycaemia is the prime consideration.
Type 2 Diabetes
Patients with type 2 diabetes are frequent participants in clinical trials, beginning with first-in-human trials. However, recruitment of these subjects can be challenging as inclusion/exclusion criteria can eliminate most study volunteers. Common reasons for excluding these volunteers include HbA1c out of range for the trial, exclusionary comorbidities, and exclusionary concomitant medications, including glucose-lowering therapies. Patients whose diabetes is managed with diet and exercise alone are frequently desired participants for first-in-human or SAD and MAD studies. However, it is often difficult to recruit these patients in the numbers needed. In the USA, while diet and exercise have conventionally been recommended as initial interventions, many patients are either commenced on metformin either at or shortly after diagnosis [78]. Potential trial participants who have subscribed to volunteer databases are frequently found to no longer be ‘naïve’ to pharmacotherapy when contacted about possible participation in a study.
For the majority of trials with patients with type 2 diabetes, the question of temporarily withdrawing routine glucose-lowering agents will be posed. Many new drug therapies to treat type 2 diabetes are being developed as add-on therapy to metformin; in such cases, the maintenance of metformin monotherapy is designed into the protocol. In circumstances when addition of an investigative drug to other ongoing oral glucose-lowering drugs is not required, patient enrolment may be accelerated by allowing washout of therapies, i.e. metformin monotherapy or washing out second-line therapies such as sulphonylureas or dipeptidyl-peptidase (DPP)-4 inhibitors. Oral glucose-lowering drugs can often be withdrawn temporarily for patients on metformin monotherapy or metformin-sulphonylurea dual therapy, with the achievement of stable blood glucose within 2 weeks [79]. These data are supported by modelling work, projecting that fasting plasma glucose concentration is stable within 30 days of washout of metformin monotherapy, sulphonylurea monotherapy, or combination of metformin and sulphonylurea-metformin dual therapy [80]. While safety parameters and glucose monitoring instructions must be built into protocols where glucose-lowering drug washout is included, if subjects are selected with HbA1c values that are less than 10 % (86 mmol/mol), the number of subjects whose fasting plasma glucose will exceed 270 mg/dL (15 mmol/L) (FDA-specified limits for trials less than 6 weeks in duration) [69] will be small, and thus, the numbers of subjects lost during washout will also be small. In addition to considering the pharmacological half-life of the drug(s) to be washed out, one should also consider the biological half-life. Most trials will exclude prior therapy with thiazolidinediones for at least 6 months because of concerns around persistence of metabolic effects long after the drug is discontinued. This reflects the genomic effects of thiazolidinediones [81].
Because other features of the metabolic syndrome are common among subjects with type 2 diabetes, issues surrounding these comorbidities should be considered. Hypertension is present in 50 % or more of individuals with type 2 diabetes [82, 83] and may or may not be well controlled at the time of screening for entry into a clinical trial. Blood pressure, if elevated, should be assessed on multiple occasions, usually after a resting period in the sitting or supine position, as individuals may be anxious when screening for study participation. An appropriate-sized cuff should be used in obese patients [84]. Patients should be questioned as to their most recent dose of medications – patients will frequently hold their medications prior to a screening visit, in spite of instructions to the contrary. Similar caveats apply when subjects check in to an inpatient unit. Generally, persistent blood pressure >160 mmHg systolic and >95 mmHg diastolic is exclusionary in most trials and should necessitate referral of the patient to a physician for further assessment and treatment.
Dyslipidaemia is another common feature of patients with type 2 diabetes [85]. This is frequently evident as increases in plasma triglycerides and decreases in HDL cholesterol compared to the nondiabetic population, although total cholesterol and low-density lipoprotein (LDL) levels are generally similar [85]. While no significant clinical effects are known for acute changes in high-density lipoprotein (HDL) cholesterol, marked elevations in triglycerides exceeding 11 mmol/L (1,000 mg/dL) are considered a risk for the development of acute pancreatitis [86] and most trials exclude patients with triglyceride levels >4.5 or 5.1 mmol/L (>400 or 450 mg/dL) if there is concern for rising triglyceride levels during the trial. Gemfibrozil, because of its potential for drug-drug interactions secondary to CYP2C8 inhibition [87], is a frequently excluded concomitant medication during metabolic trials. While LDL cholesterol concentrations are not usually elevated in patients with diabetes, the current US American College of Cardiology (ACC) and American Heart Association (AHA) guidelines recommend moderate-intensive statin therapy for all patients with diabetes between the ages of 40–75 to reduce the risk of cardiovascular events [88]. While not all trial volunteers with diabetes will be on statin therapy, many will, and careful consideration should be given to continuing these medications throughout the trial as background therapy or washing them out for subjects where statins are being used as prophylaxis. For patients with a history of known coronary heart disease, statins should not be discontinued.
Related posts:
 Regulatory Considerations for Early Clinical Development of Drugs for Diabetes, Obesity, and Cardiometabolic Disorders
Regulatory Considerations for Early Clinical Development of Drugs for Diabetes, Obesity, and Cardiometabolic Disorders
 Omics: Potential Role in Early-Phase Drug Development
Omics: Potential Role in Early-Phase Drug Development
 Assessment of β-Cell Function
Assessment of β-Cell Function
 Measurement of Energy Expenditure
Measurement of Energy Expenditure
 Assessment of Body Composition
Assessment of Body Composition
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


