Fig. 1
Telomere-DNA is an evolutionarily conserved GT-rich repetitive sequence. Basically all eukaryotes use GT-rich repetitive sequences at the ends of their chromosomes (telomeres). Despite considerable variation in telomere length and telomere-sequence, telomerase activity is the major telomere maintenance mechanism among the eukaryotes, with only few exceptions (Drosophila melanogaster). Modified from Meyne et al. (1989)
One important feature of telomeres, which is conserved in all eukaryotes, is that they posses a protruding 3′ single-stranded overhang due to the mechanism of the lagging strand DNA replication (Makarov et al. 1997; McElligott and Wellinger 1997). In mammalian cells, this single stranded overhang may fold back and invade into the preceding double stranded telomeric DNA to form a unique D-loop and T-loop structure (Griffith et al. 1999). Telomere looping may be a bona fide end protecting mechanism since it has been observed in mammals, plants and several lower eukaryotes (Cesare et al. 2003; Griffith et al. 1999; Munoz-Jordan et al. 2001; Murti and Prescott 1999). This special structure functions to seal the ends of chromosomes thus protecting them from hazardous cellular actions. In its absence, the 3′-overhang is simply occupied with specific telomere-binding proteins protecting the chromosome ends from DNA-damage.
To date a series of proteins have been described to be associated with telomeres (Fig. 2). In human cells, six proteins, TRF1, TRF2, TIN2, TPP1, RAP1 and POT1, form the shelterin complex and interact with several other proteins for telomere length regulation (de Lange 2005). Among the latter, proteins involved in DNA double-strand break repair (Ku-proteins) and non-homologous-end-joining (RAD50-NBS1-Mre11 complex) are found. It is not yet clear whether these proteins are present at the telomeres at all times or in a cell-cycle dependent manner. Among these proteins TRF1 and TRF2 form a platform for the binding and function of other telomere specific factors (Fig. 2).
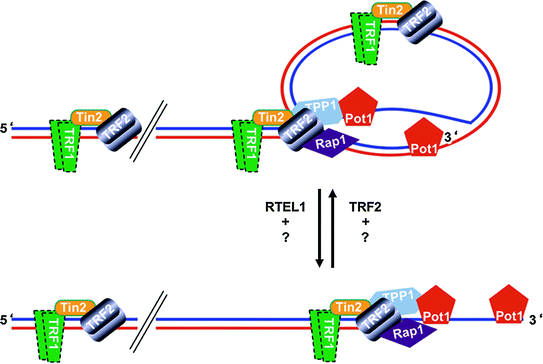
Fig. 2
Telomeres, Protein-DNA complexes at the ends of linear chromosomes, may form a lariat structure, the telomere-loop (T-loop). A schematic representation of the D-loop, T-loop structure at the chromosome ends. Lagging strand blue, leading strand red. The 3′-protruding end of the lagging strand may invade into the double stranded telomeric DNA and result in the displacement of the double strand (displacement-loop: D-loop). A large lariat structure can be observed at the telomeres (telomere-loop: T-loop). Several proteins have been localized to the telomeres. The number, temporal and spatial localization of these proteins at the telomeres is not completely understood. There is evidence that T-loop formation is facilitated by TRF2 (Doksani et al. 2013; Griffith et al. 1999) whereas RTEL1 helicase activity is required for the faithful T-loop resolution during replication (Vannier et al. 2012)
1.2 Telomerase
Telomerase is a ribonucleo-protein complex with reverse transcriptase activity with conserved sequence homology to non-LTR and LTR reverse transcriptases (Shippen-Lentz and Blackburn 1990). The activity of telomerase is necessary to overcome the ‘end replication problem’. The human telomerase enzyme is composed of two essential components, the RNA component (TERC: Telomerase RNA) which acts as a template for reverse transcription (Blasco et al. 1995); and the catalytic subunit Telomerase reverse transcriptase (TERT) with the reverse transcriptase activity (Meyerson et al. 1997; Nakamura et al. 1997). In recent years, a number of additional factors, including dyskerin, TCAB1, NOP10 and TPP1 have been identified to be constantly or transiently associated with the telomerase complex and have important functions in telomerase recruitment to telomeres or subcellular localization of the telomerase complex (Cohen et al. 2007; Collins and Mitchell 2002; Nandakumar and Cech 2013; Venteicher et al. 2009; Zhong et al. 2011; Gonzalez et al. 2014).
Telomerase is active in a variety of tumor cell lines and transformed cells in culture but not in normal fibroblasts (Morin 1989) or embryonic kidney cells (Counter et al. 1992) and most somatic human tissues do not exhibit telomerase activity (Djojosubroto et al. 2003; Kim et al. 1994; Meyerson et al. 1997; Shay and Wright 1996; Weise and Gunes 2006). In human, telomerase activity is down-regulated during embryogenesis and cellular differentiation through repression of its catalytic subunit (Gunes et al. 2000; Wright et al. 1996; Sirma et al. 2011). Due to the lack of telomerase, telomeres shorten during aging in human tissues in vivo and telomere length sets a limit to the proliferative capacity of human fibroblasts (HFs) in vitro involving the p53 and Rb pathways (Chang and Harley 1995; Harley et al. 1990; Shay et al. 1991). In this line, cells devoid of these two major pathways exhibit extended life-span but telomeres continue to shorten until a ‘crisis’ checkpoint. Cells that survive the crisis checkpoint possess telomerase activity or activate an alternative mechanism of telomere maintenance (ALT) (Counter et al. 1992). Based on these observations, Allsopp et al. (1992) proposed a model for of telomere hypothesis of ‘cell ageing and immortalization’ (Fig. 3).
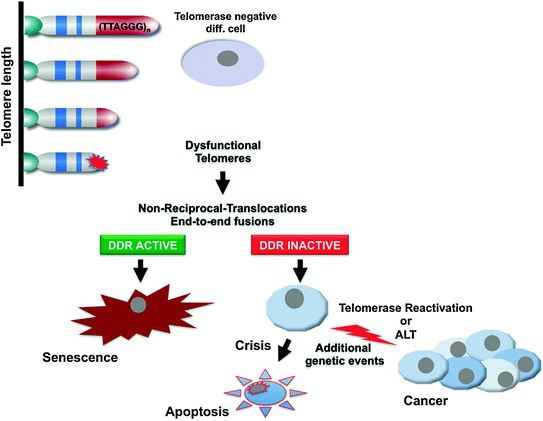
Fig. 3
Telomere hypothesis of senescence and cancer. Proliferation-dependent telomere shortening leads to telomere dysfunction, manifested by non-reciprocal-translocations and end-to-end fusions, resulting in the activation of DNA-damage checkpoints, and induction of senescence in telomerase negative, check-point proficient human cells. Checkpoint-deficient cells continue to proliferate experiencing further telomere shortening and eventually end up in crisis, characterized by apoptotic cell death, in the absence of a telomere maintenance mechanism. Activation of telomerase (or the ALT mechanism) is one of the key events to overcome crisis during tumourigenesis to stabilize telomere length and for the continuous proliferation of malignant cells
These observations together with the findings that telomerase activity can be detected in early human development but is absent in most normal somatic cells have led to the hypothesis that the down-regulation of telomerase activity in somatic cells may be a tumor-protective mechanism. In line with this hypothesis it was shown that telomerase is required for tumorigenic conversion of primary human cells (Hahn et al. 1999a). In adult human tissues some cell types maintain weak but detectable telomerase activity or telomerase activity may be induced upon stimulation. These include bone marrow stem cells, germline cells in testes, activated peripheral blood lymphocytes, skin epidermis and intestinal crypt cells (Chiu et al. 1996; Hiyama et al. 1995, 1996; Morrison et al. 1996; Ramirez et al. 1997; Ravindranath et al. 1997; Ritz et al. 2005; Weise and Gunes 2009).
Although telomerase activity could be detected in the vast majority of human cancers, it is worth mentioning that about 10–15 % of human tumors do not express detectable levels of telomerase activity. Tumors that lack telomerase activity, maintain their telomere length via a recombination-based mechanism (ALT for Alternative Lengthening of Telomeres) (Bryan et al. 1997). Experimental data indicate that telomere maintenance is required for continuous tumor cell proliferation and tumor progression (Greenberg et al. 1999; Hahn et al. 1999b; Rudolph et al. 2001). The prominent occurrence of telomerase in human cancers and data from mouse models on its requirement for tumor progression motivated the development of telomerase inhibitors to suppress tumor growth in pre-clinical studies (Damm et al. 2001; Dikmen et al. 2005; Djojosubroto et al. 2005; Herbert et al. 2002; Kumar et al. 2013; Norton et al. 1996; Zahler et al. 1991). One of these inhibitors, a lipid-conjugated 13-mer oligonucleotide that is complementary to the RNA template of telomerase, thereby directly inhibiting telomerase activity is a promising candidate and has evaluated safety, tolerability and pharmacokinetics in Phase I clinical trials. This inhibitor, Imetelstat, was developed by Geron Inc. and is now being tested to treat Hematologic Myeloid Malignancies in Phase II clinical trials. As a potential drawback, experimental studies on mouse models showed that deletion of telomerase in tumors provokes the activation of ALT as an adaptive response in cancer cells (Hu et al. 2012). It is therefore essential to explore and understand the factors that control the ALT pathway.
2 Telomere Shortening Impairs Proliferation of Transformed Cells but Dysfunctional Telomeres Can Initiate Cancer Formation
The role of telomeres in human biology was unclear until the discovery of telomerase and subsequent demonstration that telomeres shorten during aging due to the end-replication problem (Greider and Blackburn 1985; Harley et al. 1990; Hastie et al. 1990). As discussed above, telomere shortening limits the proliferation capacity of human cells, referred to as ‘Hayflick Limit’. At this stage, cells exhibit a ‘cellular senescence’ phenotype characterized by morphological changes and by the accumulation of aneuploidy, polyploidy and chromosomal fusions (Benn 1976; Saksela and Moorhead 1963; Thompson and Holliday 1975). Telomerase negative human cells that can overcome the senescence checkpoint by the expression of viral oncoproteins continue to accumulate chromosomal instability during the extended proliferation period (Counter et al. 1992). These observations indicated a pivotal role of functional telomeres in genome stability and telomerase activity thwarted telomere shortening and genomic instability (Harley 1991).
Dysfunctional telomeres can result either from alterations in the telomere-associated proteins required for end-capping function, or from alterations that promote the gradual or sudden loss of sufficient repeat sequence necessary to maintain proper telomere structure. The identification of mammalian telomerase components in the mid 90s enabled to experimentally address the functional role of telomere shortening in aging and cancer formation in vivo (Blasco et al. 1997; Rudolph et al. 1999).
Telomerase knockout mice exhibit progressive shortening of telomeres resulting in loss of telomere capping function (also referred to as telomere dysfunction) in 3rd–6th generation of knockout mice. In vivo studies supported the observations from HFs that dysfunctional telomeres are recognized by the DNA-damage-response (DDR) machinery leading to activation of p53 and Rb dependent checkpoints inhibiting tumorigenesis in cancer mouse models (Chin et al. 1999; Greenberg et al. 1999). A formal experimental prove of telomere-dysfunction induced tumor suppression in vivo was provided by studies where overexpression of c–Myc oncogene in mice with short telomeres induced genomic instability as determined by increased end-to-end fusions, non-reciprocal translocations and anaphase bridges. These genomic instability induced senescence in the presence of wild-type p53 (Feldser and Greider 2007). In fact, the tumor suppressor function was dependent on the senescence-activation function of p53 (Cosme-Blanco et al. 2007). During aging or in the absence of functional checkpoints, however, (i.e., loss of p53) or by the co-expression of oncogenic mutations, telomere dysfunction promotes genomic instability and initiates tumorigenesis (Artandi et al. 2000; Chin et al. 1999; Rudolph et al. 1999, 2001). The studies with telomerase deficient mice also underpinned the need for telomere stability—either by activating telomerase or by the ALT mechanism—for continuous tumor cell proliferation in vivo (Begus-Nahrmann et al. 2012; Ding et al. 2012; Greenberg et al. 1999; Jaskelioff et al. 2009; Rudolph et al. 2001).
In human, telomere dysfunction triggers extensive DNA fragmentation and evolution of complex chromosome abnormalities and therefore is a cancer predisposition factor (Gisselsson et al. 2001; Wu et al. 2003). The cellular basis of telomere dysfunction induced genomic instability is explained by chromosomal breakage-fusion-bridge (BFB) cycles (McClintock 1939, 1941). Persistent or transient telomere dysfunction in telomerase knockout mice can result in increased mutation rates and induce BFB-cycles resulting in gains and losses of chromosomes (Blasco et al. 1997; Hackett et al. 2001; Lee et al. 1998; Rudolph et al. 2001). Although BFB-cycles seem to be the major physiological outcome of dysfunctional telomeres, persistent telomere dysfunction can induce genomic instability via cytokinesis failure and tetraploidy (Davoli et al. 2010; Pampalona et al. 2012).
Progressive telomere shortening may also result from mutations in shelterin proteins and telomerase have been shown to be associated with human pathologies. Mutations in telomerase components (TERT, TERC, DKC1) telomerase associated factors (NOP10, NHP2, WRAP53) or the shelterin components (TRF1, TRF2, POT1) forms a bigger portion of several human diseases, like dyskeratosis congenita, aplastic anemia, pulmonary fibrosis, malignant melanoma and late stage liver cirrhosis (Hartmann et al. 2011; Savage et al. 2008; Shi et al. 2014; Vulliamy et al. 2001, 2004, 2005; Walne et al. 2007, 2008; Yamaguchi et al. 2005, 2010; Zhong et al. 2011). Mutations in telomerase components result in reduced telomerase activity and accelerated telomere shortening and thus accelerated stem cell exhaustion with age, accompanied by an increased frequency of chromosomal breaks and chromosomal aberrations and increased risk for cancer formation (Calado et al. 2012).
Together, both, mice and human studies indicate that telomere dysfunction induced genetic instability occurs through persistent bridge-breakage events, leading to a continuous reorganization of the tumor genome. These findings also show that senescence and apoptosis induced by telomere dysfunction and p53 activation contribute to tumor suppression.
3 Activation of Checkpoints as a Consequence of Telomere Dysfunction
Due to their structure and shielding by shelterin components telomeres are protected from irregular repair activities. Studies on shelterin components have identified at least six different DNA damage repair pathways that protect telomeres from irregular recombination events (Martinez et al. 2012; Sfeir and de Lange 2012). The choice of the repair pathway is dependent on the type of DNA-damage and the cell type and dictates the cellular consequences in response to telomere dysfunction. Mammalian DSBs are repaired primarily by homologous recombination (HR) or non homologous end joining (NHEJ). Gene knockout studies have revealed that loss of the shelterin components TRF1 and TRF2 activates ATM/ATR signaling for NHEJ whereas dysfunctional telomeres due to loss of POT1 trigger ATR-signaling or the activation of homologous DNA repair. Activation of the classical (c-NHEJ) or alternative (alt-NHEJ) non homologous end-joining repair pathways involving MRN complex (MRE11, NBS and Rad50), DNA-PK and Lig4 (c-NHEJ) or Lig3 or CtIP (alt-NHEJ) (Rai et al. 2010) initiate end-to-end fusions but repair activities at dysfunctional telomeres leads to chromosomal fusions, which are not stable during the cell cycle and can be a source of genetic instability (d’Adda di Fagagna et al. 2004; Takai et al. 2003). Upstream protein kinases such as ataxia telangiectasia mutated (ATM) and ATR as well as the downstream protein kinases CHK1 and CHK2 are also involved in the 5′-end-resection at dysfunctional telomeres causing a G1 cell cycle arrest or the senescence response by activating the tumour suppressor p53 pathway. In the absence of p53BP, a target of the ATM kinase that accumulates at the sides of DNA damage and suppresses end-resection, the classical NHEJ pathway is inhibited and may direct the repair mechanism towards the homologous repair, resulting in increased recombination at dysfunctional telomeres, a phenotype observed in telomerase negative, ALT-positive tumor cells (Dimitrova et al. 2008; Martinez et al. 2012).
Whether the same pathways are activated as a consequence of physiological telomere shortening remains to be shown but some data exist indicating that the alt-NHEJ is the major pathway to repair DNA damage at naturally occurring dysfunctional telomeres (Rai et al. 2010). The p16/INK4a-Rb pathway has been implemented to contribute to the detection of telomere-induced DNA damage, activating the senescence pathway and recent data show that p16/INK4a protects cells against dysfunctional telomere–induced ATR-dependent DDR in Pot1b deficient mice but the contribution of p16 remains still elusive yet (Shay et al. 1991; Wang et al. 2013). Elucidating the DDR pathways in response to physiological telomere dysfunction would be crucial to better understand the role of genomic instability to tumorigenesis during aging.
4 Telomere-Dysfunction and Induction of Senescence as a Tumor Suppressor Mechanism
As discussed above, telomere shortening is regarded as the main cause of telomere dysfunction leading to induction of replicative senescence in aging cells. There is now emerging evidence that the accumulation of telomeric DNA damage in response to DNA replication stress can also contribute to induction of senescence. The induction of this checkpoint involves abrupt induction of replication stress at telomeres, which appears to be independent of classical telomere shortening (Fig. 4).
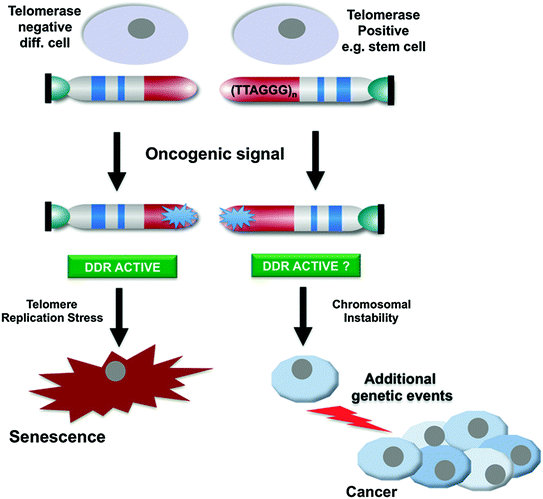
Fig. 4
Telomerase activity alleviates telomere replication stress and facilitates to overcome oncogene-induced senescence. Oncogene activation leads to abrupt accumulation of DNA damage at telomeres resulting in senescence and tumour suppression. Telomerase-positive stem cells could be resistant to oncogene-induced senescence and may be selected as the cell type of origin of tumour development
Dysfunctional telomeres can be detected by the accumulation of telomere dysfunction-induced foci (TIF) at the telomeres (d’Adda di Fagagna et al. 2003; Takai et al. 2003). These foci include 53BP1 and phosphorylated H2AX (gamma-H2AX) at the dysfunctional telomeres. Interestingly, recent observations show the accumulation of persistent TIFs upon oncogene-induced senescence (OIS) or stress-induced senescence (Fumagalli et al. 2012; Hewitt et al. 2012; Suram et al. 2012). We recently showed that aneuploidy-induced senescence (AIS) involves replication stress and TIF formation at telomeres indicating that telomeres seem to mediate (AIS) (Meena et al. 2015). These new findings may provide a unifying mechanism for senescence as a general tumor suppressor mechanism whereby telomeres may converge different kinds of cellular stress in one pathway (Reviewed in Gunes and Rudolph 2012, 2013).
The biological basis for this function of telomeres as a sensor of replication defects may be due to their specific sequence composition and structure. Telomeres can form G-quadruplex structures (G4) by intra-molecular Hoogsteen G-G base pairs. G4 structures increase in a cell cycle dependent manner in human cells (Biffi et al. 2013) and preferentially form at the 3′-end of chromosomes (Tang et al. 2008), are highly stable. G4 structures are thought difficult to resolve during replication and may provoke replication fork stalling and chromosome fragility (Tarsounas and Tijsterman 2013). Fragile sites are particularly prone to chromosomal breakage and recombination events as a result of replication stress (O’Keefe and Richards 2006). Replication stress can be induced by inappropriate proliferation signaling such as oncogene activation or loss of cell cycle inhibitors that deregulate transcription and generate DNA damage (Bermejo et al. 2012; Di Micco et al. 2006). Telomeres are difficult to replicate and may lead to fork stalling during replication upon inflated proliferation signals (Suram et al. 2012). Consistently, replication stress at telomeres and thus inefficient replication of telomeric DNA could attract DDR and induce the senescence checkpoints as a tumor suppressor mechanism. In cells defective in functional repair mechanisms or faithful telomere replication, however, dysfunctional telomeres can initiate genome instability.
5 TRF1 and Telomerase in the Context of Telomere Replication Stress
There is emerging experimental evidence that replication through difficult replicating sites requires coordinated action of telomerase activity, telomere binding proteins and specific helicases that are recruited to the telomeres for faithful replication. TRF1 plays a key role in this context. Loss of mammalian TRF1 or its fission yeast counterpart Taz1 leads to stalled replication forks and fragile telomere phenotype (Martinez et al. 2009; Miller et al. 2006; Sfeir et al. 2009). Importantly, MEFs from TRF1 deficient mice exhibited a premature senescence phenotype compared to their wild type counterparts; in the absence of cellular checkpoints, i.e., in cells expressing SV40-LT, the senescence phenotype was rescued but led to increased chromosomal instability (Martinez et al. 2009). At organismal level, mice lacking TRF1 in the stratified epithelia (TRF1flox/flox × K5-Cre transgenic bitransgenic mice) showed dysfunctional telomeres associated with skin hyperpigmentation and epithelial dysplasia but died perinatally. When these mice were crossed with p53 null mice, they could survive but exhibited an increase in squamous cell carcinoma. Together, these studies indicate that telomere replication is facilitated by the shelterin factor TRF1 to prevent replication fork stalling and that telomeric replication stress generates fragile telomeres that can instigate genomic instability and cancer.
Interestingly, BLM helicase, which is also able to bind and resolve G4 structures, interacts with TRF1 and is recruited to telomeres during replication in late S/G2 and cells lacking BLM accumulate dysfunctional telomeres and telomere-dependent chromosome fusions (Barefield and Karlseder 2012). RTEL1 is another helicase that facilitates faithful telomere replication, potentially by resolving the G-quadruplex structures at the T-loop (Vannier et al. 2012, 2013). Other helicases with G4 resolving activity include the recQ helicases WRN, RECQL4 and DNA2. DNA2 deficiency results in defective telomere replication, leading to elevated fragile telomeres, telomeres loss, and telomere DNA damage response (Lin et al. 2013). In the same line, it has recently been demonstrated that the activity of the Pif1 helicase, that can associate with telomerase, is required to open telomeric G4 structures and that the enzymatic activity of telomerase is crucial for this function indicating that the damage present at telomeres is repaired by telomerase (Chang et al. 2009; Mateyak and Zakian 2006; Paeschke et al. 2011). It remains speculative whether Pif1 activity precedes and facilitates telomere replication or it is required to resolve structures generated during replication. Studies in the ciliate Stylonychia lemnae indicate that telomerase recruitment by the telomere binding protein-ß, the homologue of the mammalian shelterin protein TPP1, facilitates unfolding G4-structures. However, the exact mechanisms how these helicases act to resolve telomeric G4 and their differential functions remain elusive.
Recent studies indicate that BRCA2 and RAD51 act in concert to heal fragile telomeres in mouse cells, probably by enabling the restart of replication at stalled replication forks that are processed by HR during the S-phase (Badie et al. 2010). BRCA2 recruits RAD51 to the telomeres during replication in S-phase and both factors are required for maintenance of telomere length in mouse embryonic fibroblasts (MEFs). Consistently, MEFs lacking BRCA2 or RAD51 exhibited an increased fragility, telomere shortening and telomere dysfunction induced DNA damage foci (TIF) indicative of loss of telomere protection. Interestingly, telomerase positive cells showed higher fragility in the context of BRCA2 mice when compared to telomerase negative cells with shorter telomeres from late generation telomerase knockout cells. This result indicates that longer telomeres have a greater chance to accumulate fragile telomeres in the absence of repair mechanisms and in the presence of telomerase. In conclusion, the adult stem cells, the main cell type that retains telomerase activity in adult human tissues may represent the cell type of origin of cancer formation (Fig. 4).
Together, telomeres have a dual role in cancer formation. Telomere shortening and telomere replication stress in malignant cell clones serve as a tumor suppressor mechanism by activating senescence and or crisis checkpoints. In contrast, telomere shortening in aging tissues can also lead to an induction of chromosomal instability by promoting chromosomal fusion and fusion-bridge-breakage cycles. In addition, the inhibition of cell proliferation in aging tissues can also increase the selective pressure for clonal outgrowth of (pre-) malignant cell clones by changing the tissue environment and by impairing proliferative competition of non-transformed cells (Bilousova et al. 2005; Braig et al. 2014; Ju and Rudolph 2006). The influence of telomeres on tumor protection/tumor promotion may depend on the lifetime. Early in life when telomeres are long cancer protective effects of telomere shortening/replication stress in malignant cell clones may be dominant. In contrast, tumor-promoting effects of telomere shortening may become dominant in aged tissue and tissues experiencing telomere shortening in response to chronic diseases such as liver cirrhosis in response to hepatitis or progressive stages of ulcerative colitis (Rabinovitch et al. 1999; Rudolph et al. 2009). It remains to be investigated whether targeting of senescence checkpoints in response to telomere shortening or telomere replication stress could lead to development of novel anti-cancer therapies and how these approaches affect tissue aging. Studies in mouse models indicate that it is possible to improve tissue maintenance without increasing cancer risk by inhibiting downstream checkpoint responses (Cdkn1a/p21) that limit proliferation of cells in response to telomere shortening (Choudhury et al. 2007). In addition, it was shown that p21 deletion can have anti-tumor effects in mouse models of leukemia or irradiated human tumor cells (Lazzarini et al. 2008; Viale et al. 2009; Waldman et al. 1996). It is possible that the tumor inhibiting effects of p21 deletion involve the increase in telomere replication stress in genomically instable tumor cells. Together, these studies suggest that it should be possible to define molecular targets that can improve both tissue maintenance and cancer protection in aging tissues.
Important areas of future research include the delineation of (i) distinct cellular stress factors that cause telomere replication stress, (ii) molecular mechanisms that are involved in the induction of replication stress, (iii) activation of checkpoints in response to replication stress at telomeres, and (iv) mechanism how telomerase contributes to the suppression of telomere replication stress.
References
Allshire RC, Dempster M, Hastie ND (1989) Human telomeres contain at least three types of G-rich repeat distributed non-randomly. Nucleic Acids Res 17:4611–4627PubMedCentralPubMed
Related posts:
 Modelling: What Have Mouse Models for Chromosome Instability Taught Us?
Modelling: What Have Mouse Models for Chromosome Instability Taught Us?
 of Chromosomal Instability
of Chromosomal Instability
 of Aneuploidy in Cancer: Transcriptome and Beyond
of Aneuploidy in Cancer: Transcriptome and Beyond
 Diverse Effects of Complex Chromosome Rearrangements and Chromothripsis in Cancer Development
Diverse Effects of Complex Chromosome Rearrangements and Chromothripsis in Cancer Development
 of Chromosomal Aberrations in Solid Tumors
of Chromosomal Aberrations in Solid Tumors
 as Models of Mitotic Fidelity
as Models of Mitotic Fidelity
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


