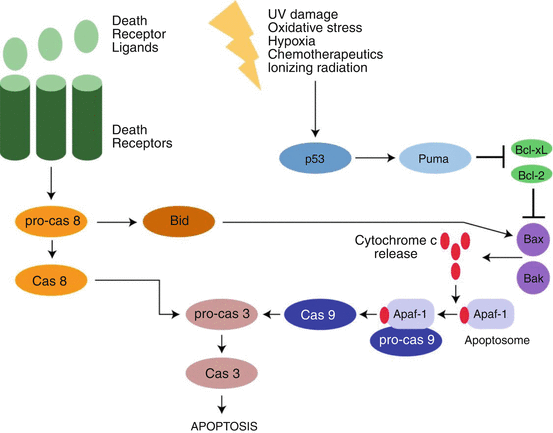
The molecular and genetic basis of apoptosis has been extensively explored. Defining programmed cell death by biochemical processes rather than morphological changes allows better elucidation of differences in processes, because the end result of many different biochemical processes can look the same. Based on a molecular–mechanistic approach, apoptosis can be further divided into extrinsic apoptosis and intrinsic apoptosis. Figure 2.2 from (Vucic et al. 2011) shows a model of the intrinsic and extrinsic pathways. The extrinsic pathway initiates cell death by activation of cell surface receptors. The surface receptors that initiate the process are called death receptors, and these include the receptors for CD95 (Fas/Apo1), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), and tumor necrosis factor alpha (TNFα). These receptors all have a trans-cytoplasmic “death domain” that transmits the signal from the extracellular milieu to inside of the cell (Ashkenazi and Dixit 1998). Binding of the cognate ligand (e.g., TNFα with the surface membrane TNFα receptor) activates a process leading to the formation of the death-inducing signaling complex (DISC). The DISC then initiates the activation of procaspase 8. Caspases are aspartic proteases that require the presence of a cysteine to be active (i.e., cysteine-dependent aspartate-specific protease, or caspase). Caspases are primarily involved in two processes – apoptosis and inflammation (McIlwain et al. 2013). Caspases 3, 6, 7, 8, and 9 are involved in apoptosis, and 1, 4, 5, and 12 in inflammation. Because the caspases involved in apoptosis will lead to cell death, when not involved in apoptosis they are in the cell in the inactive state as procaspases.
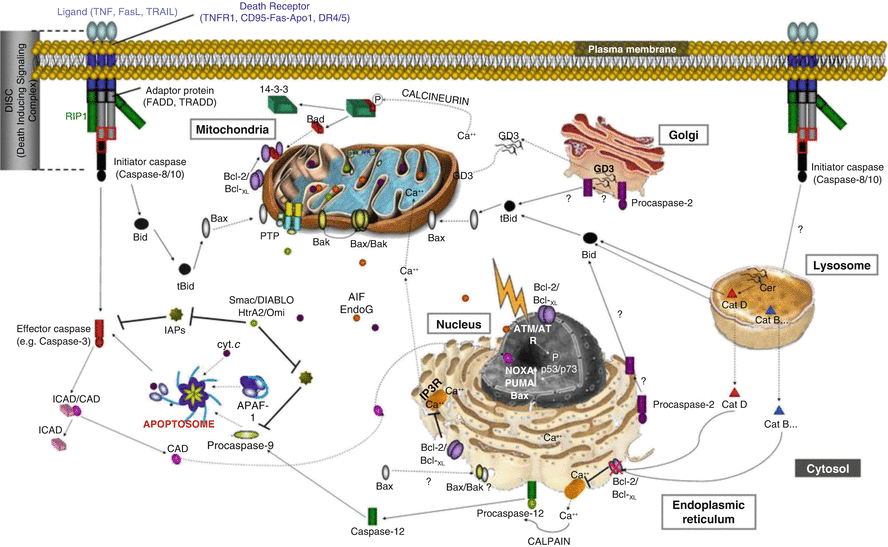
The extrinsic apoptosis process continues with the conversion and activation of procaspase 8 to caspase 8 by dimerization and cleavage of the procaspase. Caspase 8 is an initiator caspase, and it then converts the procaspases 3 and 7 into the executioner caspases 3 and 7, which then initiate the apoptosis process and cell death. Apoptosis is a rapid process, and leads to the elimination of the cell often in less than 24 h. As noted previously, this process occurs without developing an inflammatory response. The caspase mechanisms in apoptosis are shown in Fig. 2.3.
The other mechanism of apoptosis is termed intrinsic apoptosis and is induced by intracellular stress or damage. It can also occur during normal development. Intrinsic apoptosis starts when caspase 9 is activated. This occurs when a stress causes the mitochondria to release cytochrome c, and the cytochrome c then binds with the adapter protein, apoptotic protease activating factor-1 (APAF1). Seven activated APAF-1 molecules assemble with caspase 9 monomers and cytochrome c to activate the caspase 9 by dimerization. The complex of the cytochrome c, APAF-1, and active caspase 9 is called the apoptosome (McIlwain et al. 2013). The active caspase 9 then activates caspase 3 and enters into the same executioner phase as the extrinsic apoptosis process.
Intrinsic apoptosis is not receptor-initiated, but is controlled by stimuli that change the internal milieu (Elmore 2007). For example, the absence of growth factors can cause a failure of the suppression of intrinsic apoptosis and therefore the start of the death cascade. The cascade can be stimulated by stresses such as hypoxia and by cell-damaging agents such as radiation and free radicals. Whatever might be the agent, there is induction of changes in the mitochondrial membrane which opens the mitochondrial permeability transition (MPT) pore which then allows the release of cytochrome c into the cytosol (as described above). Mitochondrial membrane integrity is regulated by the Bcl-2 family of proteins. These can be pro-apoptotic or anti-apoptotic. Among the Bcl-2 proteins that inhibit apoptosis are Bcl-2, Bxl-2, Bcl-w, Mcl-1, and Bcl2L10. Those that promote apoptosis include Bax, Bak, Bok, Bid, Bad, Noxa, Puma, and Bim (Arya and White 2015). This will be discussed as part of the role of apoptosis in cancer treatment.
2.2.2 Autophagy
The second major form of programmed cell death is autophagy. The majority of research on autophagy has been on yeast, but similar pathways are in mammalian cells. There are three forms of autophagy: microautophagy, macroautophagy, and chaperone-mediated autophagy. In all the three cases, the goal of autophagy is the proteolysis of cytosolic contents by lysozymes. In microautophagy, this occurs by direct incorporation of the material into lysosomes through membrane invagination. In chaperone-assisted autophagy, the material is directed across the lysosomal membrane by a protein complex. In macroautophagy, the material to be degraded is first encased in a membrane and then degraded after this complex fuses with a lysosome (Glick et al. 2010). This latter process, macroautophagy, will be discussed here and will be called by the generic term “autophagy.” Autophagy is initiated when there is a stress on a cell such as starvation or hypoxia. On the morphological level, autophagy is characterized by degenerating cytoplasmic material inside double-membraned vesicles called autophagosomes, which then merge with lysosomes to form autolysosomes. Within the autolysosome, the cellular debris and the inner membrane of the autolysosome are dissolved by the acidic lysosomal hydrolases (Kroemer et al. 2009).
As mentioned previously, autophagy has been primarily studied in yeast. Autophagy in mammals is not as well elucidated. Macroautophagy (again, generally referred to as autophagy) is nonspecific, with the cellular debris swept randomly into the autophagosomes. The cause of the formation of the autophagosome membranes in man is unknown. In yeast, the phagophore membrane is formed around the preautophagosomal structure (PAS), but a similar structure has not been found in mammals (Glick et al. 2010). The primary proteins involved in autophagy in yeast are the Atg proteins. The homologs in mammals are called the ATG proteins. The primary Atg protein is the Atg1 kinase complex. The homolog in mammals is the ULK1/2 complex (Feng et al. 2014).
Selective autophagy is similar to macroautophagy, except that there is receptor-mediated binding of specific compounds into the autophagosomes rather than random debris. An example is mitophagy, in which senescent mitochondria are degraded by autophagy (Youle and Narendra 2011). It is uncertain if autophagy is an important form of cell death. Although autophagosomes are seen in dying cells, they may represent failed protection of the cells rather than a mode of death of the cells (Fuchs and Steller 2015). A review of 1400 active or potential anticancer agents showed that none caused autophagic cell death, although signs of autophagy could be found. Further, elimination of genes necessary for autophagy, such as ATG5 and ATG7, accelerated rather than inhibited cell death (Shen et al. 2012).
2.2.3 Necrosis
The third major classification of programmed cell death is death by necrosis. This was the last to be understood to be under the control of molecular mechanisms. Historically, it was thought that necrosis was a passive form of cell death, similar to Aristotle’s death by violence (Ziegler and Groscurth 2004). It is now recognized that there are distinct regulatory proteins that can be involved in necrosis (Feoktistova and Leverkus 2015). The morphological changes of necrosis include increased cell volume, swelling of the cellular organelles, and then rupture of the cell membrane with cell death (Kroemer et al. 2009). The most studied form of programmed necrosis is necroptosis. In necroptosis, TNF-1 stimulates the TNF receptor 1 (TNFR1). The receptor-interacting protein kinase 1 (RIPK1) then binds with the TNFR1-associated death domain (TRADD) to form the RIPK1- and TRADD-dependent receptor-bound complex 1. This complex then activates nuclear factor-kB (NF-kB), which causes inhibition of apoptosis and ultimately the formation of a cytosolic death-inducing signaling complex (DISC) and a cascade that results in regulated necrosis and cell death (Vanden Berghe et al. 2014). Necroptosis is defined as necrosis involving activation of RIPK1 and RIPK. The RIPK1–RIPK3 complex is called the necrosome (Feoktistova and Leverkus 2015). Much of the pathway leading from TNF stimulation of the cell surface membrane death receptors (discussed above in relation to apoptosis) through to necroptosis is shared with apoptosis, and necroptosis primarily occurs when the apoptotic pathway is inhibited (Vanlangenakker et al. 2012). Necroptosis can also be stimulated by viruses such as human immunodeficiency virus type 1 (HIV-1), herpes simplex virus type-1 (HSV-1), the West Nile virus (WNV), and the murine cytomegalovirus (MCMV). Bacterial infections, such as Shigella flexneri, Neisseria gonorrhoeae, and Klebsiella pneumoniae, can also stimulate necroptosis. Physicochemical stressors such as radiation and chemotherapy can also induce necroptosis (Vanlangenakker et al. 2012). There are other forms of programmed necrosis using other pathways to a similar morphological endpoint. These include ferroptosis, oxytosis, ETosis, NETosis, cyclophilin D-mediated necrosis, parthanatos, pyroptosis, and pyronecrosis (see (Vanden Berghe et al. 2014) for a review).
2.3 Phagosomes and Lysosomes and Cell Content Degradation
All of the programmed and necrotic cell death mechanisms ultimately must degrade the components of the cells back into their original building blocks, such as fats, amino acids, and nucleic acids. This process is accomplished by phagosomes and lysosomes. Phagocytes are cells that can ingest other cells and cellular debris. Whereas the elimination of a pathogen, such as bacteria, can induce an immune response, clearance of the products of apoptosis usually induces no immune response. One suggestion is to call the latter process “efferocytosis,” based on the Latin term “efferre” meaning “to take to the grave” or “to bury” (Henson and Hume 2006). This process can be broken down into four steps: the dying cell sends out a “find me” signal; then the phagosome finds and recognizes the dying cell; next the phagocytes engulf the dying cell; and finally the phagosome digests the cell (Ravichandran and Lorenz 2007). The “find me” signals from the dying cell include the nucleotides ATP and UTP, the chemokine fractalkine (CX3CL1), and the lipids lysophosphatidylcholine (LPCAT1) and sphingosine-1-phosphate (S1P, lysosphingolipid) (Arandjelovic and Ravichandran 2015). The apoptotic cell presents a surface signal on its surface, such as the lipid phosphatidylserine (PtdSer). In healthy cells, this lipid is only on the inner side of the membrane, and it is exposed in apoptotic cells (Balsubramanian and Schroit 2003). Healthy cells can also avoid engulfment with protective surface signals, including CD47 and CD31 (Arandjelovic and Ravichandran 2015). Once the phagosome has identified the cell to be degraded, it then starts the engulfment process. This engulfment is macroautophagy, as described above, in which an autophagosome is formed. This starts with the formation of the phagophore membrane, probably from multiple sources including the Golgi complex, the endoplasmic reticulum, and mitochondria. The membrane then grows circularly around the contents to be engulfed (such as cells or organelles) and ultimately forms a complete membrane around the cargo, which is called an autophagosome. The autophagosome can then fuse with other endosomes containing debris to be degraded. Finally, the autophagosome fuses with a lysosome (or lysosomes) for the final processing of the cargo (Mijaljica et al. 2012).
Lysosomes are single-membrane vesicles that contain hydrolytic enzymes to degrade cell contents (Boya 2012). The enzymes within the lysomes are only active at acidic pH, and ATPase proton pumps maintain an intra-vacuole pH of 4.6–5.0 (Mellman et al. 1986). The mechanism of fusion of the lysosomes with the autophagosomes is not completely elucidated. The current belief, based on time-lapse focal microscopy, is that there is a combination of fusion of the two organelles and of “kissing” in which there is contact and transfer of the autophagosome contents into the lysosome for degradation (Luzio et al. 2007). There, the cellular contents are degraded and then transported back to the cytosol for reuse.
Lysosomes are also involved in apoptosis (Mrschtik and Ryan 2015). Induction of lysosomal membrane permeation, for example, through proteins accumulated in the lysosomal membrane or from reactive oxidative species generated within the lysosome, can cause release of lysosomal enzymes. The enzymes implicated in lysosomal induction of apoptosis include the aspartic cathepsin D and the cysteine cathepsins B, C, F, H, K, L, O, S, V, W, and X (Mrschtik and Ryan 2015). Cathepsin D may be directly involved in apoptosis initiation through interaction with the pro-apoptotic Bcl-2 proteins Bax and Bid. Other studies suggest that the released lysosomal enzymes may not initiate apoptosis but can enhance it (Oberle et al. 2010).
2.4 Programmed Cell Death and Cancer
A cancer cell can be described as a cell that has malfunctioning normal cell death mechanisms. That is, programmed cell death, primarily apoptosis, is not active, allowing the cells to have unchecked growth. Autophagy is also malfunctioning, both in allowing the abnormality to occur rather than removing it and in allowing the cells to survive. The normalization of programmed cell death is therefore a prime target for the treatment of cancer. As stated earlier, cell death is perhaps the only area of cancer where death is welcomed.
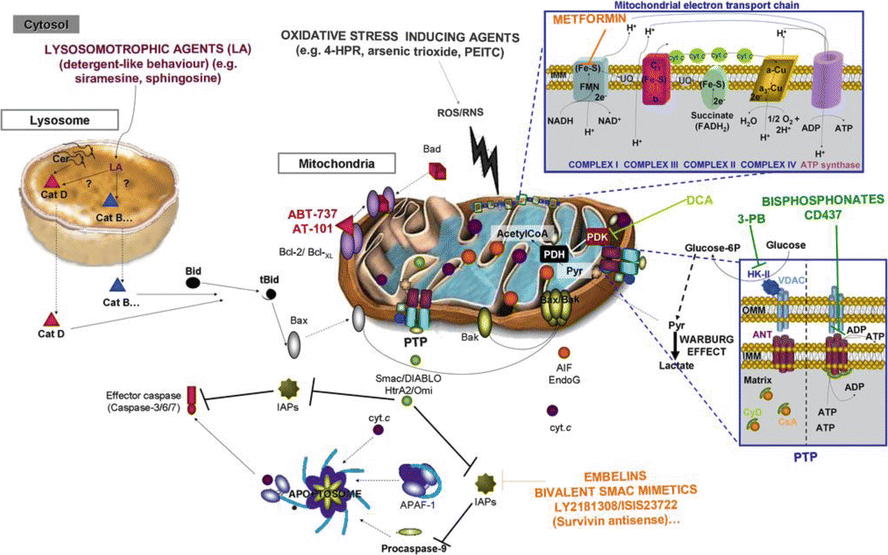
Resistance to apoptosis can be seen in carcinogenesis, tumor progression, and the development of resistance to treatment (Mohammad et al. 2015). Apoptotic activity is a function of balance between the pro-apoptotic proteins and the anti-apoptotic proteins. Disruption of this normal balance toward the anti-apoptotic proteins can encourage cancer cell growth (Kang and Reynolds 2009). For example, the Bcl-2 family contains both pro-apoptotic and anti-apoptotic members. Pro-apoptotic Bcl-2 proteins such as BAX and BAK are required for activation of the mitochondrial outer membrane permeabilization (MOMP) process of the intrinsic apoptotic pathway. These two proteins can be stimulated by BIM, BID BAD, and PUMA. They are inhibited by Bcl-2, BCL-X, Bcl-2A1, BCL-W, and MCL-1. The overexpression of anti-apoptotic proteins can be found in different cancer clones. Some cancer cells are “Bcl-2 dependent” for survival, and as such are dependent on Bcl-2 or Bcl-2-like proteins for apoptotic inhibition and growth (Juin et al. 2013). Figure 2.4 highlights the potential therapeutic intervention sites in the apoptosis pathway.
A potentially efficacious therapeutic modality is to activate apoptosis and cancer cell death through manipulation of the Bcl-2 interactions. This has been explored in the laboratory and in the clinic. One approach is with BH3 mimetics. The BH3 domain is a sequence of 15 amino acids that binds to a hydrophobic groove in the anti-apoptotic proteins of the Bcl-2 family, thereby inhibiting their anti-apoptotic activity and activating apoptosis. At least 20 compounds have been explored as BH3 mimetics to stimulate apoptosis (see the review by Vela and Marzo (2015)). Of these, the two most studied are the natural product gossypol and ABT-737.
Gossypol is extracted from cottonseed oil. Along with being explored for use to treat cancer, it is also being looked at as a male contraceptive, to lower cholesterol, and as an antibacterial and antiviral agent (Keshmiri-Neghab and Goliaei 2014). Gossypol is a polyphenolic bis-sesquiterpene that is extracted from cottonseed oil as a racemic mixture. Both the natural racemic mixture and the more active R-(-) enantiomer (AT-101) have been explored for cancer treatment. Gossypol has multiple activities, including inhibiting DNA replication and repair, inhibiting cell cycling, as well as inducing apoptosis (Keshmiri-Neghab and Goliaei 2014). The mechanisms inhibiting apoptosis include inactivation of cMyc, post-translational modification of the Akt, and downregulation of inhibitors of apoptosis, including inhibitors of apoptosis (IAPs). Gossypol also induces release of cytochrome c from mitochondria, thereby initiating the intrinsic apoptosis pathway, and acts as a BH3 mimetic and induces apoptosis. So far in Phase II clinical trials, AT-101 was found to be tolerable to patients but showed no significant survival advantage in prostate cancer, small cell lung cancer, or nonsmall cell lung cancer (Baggstrom et al. 2011; Ready et al. 2011; Sonpavde et al. 2012).
There is also extensive preclinical research on the Bcl-2 mimetic ABT-737 and its oral form ABT-263 (navitoclax). ABT-737 binds to Bcl-2, BCL-XL, and BCL-W. In-vitro ABT-737 induces apoptosis in a wide variety of tumor cell lines (Billard 2013). Single agent navitoclax was active in a Phase I study of leukemia (Roberts et al. 2012) and not active as a single agent in a Phase II study in relapsed small cell lung cancer (Rudin et al. 2012).
An alternative approach to modulating Bcl-2 overexpression is to inhibit the synthesis of Bcl-2. Oblimersen sodium is an 18 mer antisense oligonucleotide that targets the first six codons of Bcl-2 RNA. It attaches to the Bcl-2 RNA and inhibits its transcription (No author listed 2007). Preclinical research suggested that oblimersen is synergistic with chemotherapy (Moreira et al. 2006). A randomized Phase III trial for high-risk melanoma patients of dacarbazine with or without oblimersen showed no survival advantage for the oblimersen (Bedikian et al. 2014). A randomized trial of fludarabine and cyclophosphamide with or without oblimersen for relapsed or refractory chronic lymphocytic leukemia showed no overall advantage with oblimersen, but there was a higher response rate, and for patients who responded to treatment there was a significant increase in 5-year survival (O’Brien et al. 2009). A trial of dexamethasone with or without oblimersen for patients with refractory or relapsed advanced multiple myeloma showed no time to progression or response advantage with oblimersen (Chanan-Khan et al. 2009). A trial for castration-resistant prostate cancer of docetaxel with or without oblimersen showed no increase in response rate with oblimersen. Similarly, a randomized study of patients with extensive staged small cell lung cancer comparing carboplatin and etoposide with or without oblimersen did not show any outcome improved by oblimersen (Rudin et al. 2008).
Myeloid cell leukemia-1 (Mcl-1) is a prosurvival Bcl-2 protein which is overexpressed in many cancers (Akgul 2009). Inhibition of Mcl-1 sensitizes cancer cells to both chemotherapy and radiation therapy (Warr and Shore 2008). The levels of Mcl-1 in cells can be increased due to stimulation of transcription of Mcl-1, as seen in myeloma cells. In myeloma cells, the upregulation involves the JAK/STAT pathway which induces IL-6 and IFN-α signaling and Mcl-1 transcription (Puthier et al. 2001). Mcl-1 translation is under the control of the micro-RNA mir-29b (Mott et al. 2007). Mcl-1 has a short post-translation lifetime and is degraded through ubiquitin-dependent protein degradation by the 26S proteasome (Warr and Shore 2008). Inhibition of the degradation leads to inhibition of apoptosis and protection from cisplatin and etoposide treatments (Zhong et al. 2005). Mcl-1 dependence was shown to predict outcome to treatment of patients with acute myeloid leukemia receiving vorinostat and gemtuzumab ozogamicin (Pierceall et al. 2014). Similarly, Mcl-1 expression predicts progression-free survival in patients with chronic lymphocytic leukemia treated with pentostatin, cyclophosphamide, and rituximab (Awan et al. 2009). As of yet, no intervention has been tested that is designed to increase Mcl-1 degradation and allow increased apoptosis.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


