Topoisomerase IIα
Topoisomerase IIβ
Unlinks tangled daughter chromosomes after replication and resolves DNA knots during recombination. Essential for survival of proliferating cells and its levels rise during cell growth. Essential for chromosome condensation, cohesion, segregation, and remodeling
It is known to play important role in transcription of hormonally or developmentally regulated genes
Its concentration peaks at G2/M phase. Accumulates ahead of replication forks and transcription complexes
Its concentration is independent of cell cycle, and it is found in high levels in all cell types regardless of proliferation status
The gene encoding α form is localized to chromosome 17
The gene encoding β form is localized on chromosome 3
It is found on chromosomes during mitosis
It dissociates from the chromosomes during mitosis
The physiological regulation of topoisomerase II is important for cell cycle homeostasis (Wang 2002), and its mechanism of action is evidenced by various reports (Capranico and Binaschi 1998; Tse et al. 1980; Deweese et al. 2008; Schoeffler and Berger 2005; Nitiss 2009a; Champoux 2001; Schoeffler and Berger 2005). Topoisomerase II needs Mg2+ and ATP for its catalytic cycle (Mc Clendona and Osheroff 2007) and involves a two-gate mechanism (Roca and Wang 1994). The reaction cycle of mechanism of action of topoisomerase II is shown in Fig. 20.1.
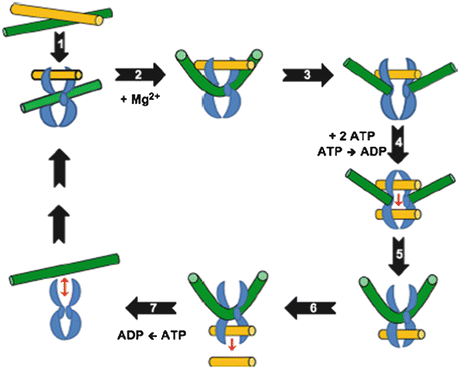
Fig. 20.1
Catalytic cycle of topoisomerase II (Adapted from Ketron and Osheroff (2014). The homodimeric enzyme is shown in blue, the DNA double helix that is cleaved and acts as the DNA gate (G-segment) is shown in green, and double helix that is transported through DNA gate (T-segment) is shown in yellow. Topoisomerase II binds non-covalently to the first segment called as the gate (G) duplex that will be cleaved. G duplex–topoisomerase II complex binds at a crossover region with transported (T) duplex. ATP binds and promotes formation of topological complex followed by hydrolysis of one of the bound ATP molecules. Mg2+-dependent cleavage of G duplex follows. DNA is sampled for malleability and sequences that can be cleaved are bent to an angle of 150°. T duplex passes through a gap formed in G duplex and T segment is released through C-terminal protein gate. G duplex is religated and bound ATP is hydrolyzed. Enzyme is now free to initiate another catalytic cycle
The level of topoisomerase II is increased by many folds during normal cell growth and also in tumor cells which is reported in many studies (Hsiang et al. 1988; Woessner et al. 1991), but in tumor cells, this increase continues which can lead to uncontrolled growth resulting in cancer. The regulation of topoisomerase II activity is a complex process (Isaacs et al. 1998). It is important to find out the exact mechanism of action of topoisomerase II inhibitors and how they interfere with physiological regulation of its expression to kill the cancer cells. These anticancer agents ultimately alter the functions of topoisomerase II either by acting as its catalytic inhibitors or by increasing its activity resulting into stabilization of DNA-topoisomerase II cleavage complexes in case of topoisomerase II poisons (Wilstermann and Osheroff 2003). This property of antineoplastic drugs is responsible for triggering the cell death pathways in cancer cells (Fortune and Osheroff 2000).
20.2 Topoisomerase II: A Marker for Cell Proliferation
The topoisomerase II expression serves as specific marker for elevated cell growth. In a study done with the help of a monoclonal antibody probe against topoisomerase II, the blood lymphocytes of chick erythrocytes expressed high levels of topoisomerase II de novo when they attained proliferation (Heck and Earnshaw 1986). In another study, the evaluation of different markers including topoisomerase IIα in breast cancer patients, a positive correlation was found between pathological rate and overexpression of topoisomerase IIα (Petita et al. 2004). A study in human anaplastic astrocytoma, a type of brain cancer, has also shown the significance of topoisomerase II as a reliable cell proliferation marker (Habberstad et al. 2011). There are many other experimental records with different types of cancer cells in which the detection of topoisomerase II levels has been carried out (Wilson et al. 2001; Hsiang et al. 1988). Thus, it can be concluded that therapeutic drugs may act on topoisomerase IIα as their target to decrease its expression in tumor cells but its relatively low levels in some cancer cells are correlated with drug resistance of those cells (Lynch et al. 1997). In a study, the levels of topoisomerase IIβ mRNA in HL-60/MX2 cells were found to be ≤1 % as compared to HL-60 cells (Harker et al. 1995). The expression of topoisomerases has been studied by introducing the gene of these enzymes from human cells to microorganisms such as yeast and E. coli and by genetic complementation experiments that help to find the mechanistic studies about drug–enzyme interactions (Bjornsti et al. 1989; Shiozaki and Yanagida 1991; Caron et al. 1994; Jensen et al. 1996). A study has shown that many cancer-related genes are more or less similar in yeast and humans (Foury 1997). In this way, the functional and regulational aspects of human topoisomerase II can be easily studied (Kranz and Holm 1990). A human topoisomerase IIβ construct was complemented to topoisomerase II mutations in Saccharomyces cerevisiae in a study to elucidate the use of some known as well as novel drugs against human topoisomerase IIβ in cancer cell lines (Fisher et al. 1997). These studies have been helpful in studying the exact mechanism of action of some cytotoxic drugs that interact with topoisomerase II. A study has shown that HCC1937 breast cancer cells lost their resistivity to etoposide, a known topoisomerase II poison, after genetic complementation of BRCA (gene involved in DNA repair of inter strand cross-links)-deficient cells (Treszezamsky et al. 2007). The gene sequence of topoisomerase II is reported to be consensus among different organisms and in case of some tumors (Austin et al. 1990). The presence of consensus sequences of topoisomerase II are at break points of COL4A5 and COL4A6 gene mutation in case of diffuse esophageal leiomyomatosis, a benign smooth muscle cell tumor (Ueki et al. 1998). In a recent study, the t(7;21)(p22;q22) translocation was confirmed in case of acute myeloid leukemia with the help of RT-PCR along with the detection of topoisomerase II sequence near break point (Giguere and Hébert 2011).
20.3 Catalytic Inhibitors of Topoisomerase II
Recently, attention has been focused on phytochemicals (the non-nutritive components in plant-based diet) that possess cancer-preventive properties (Surh 2003). Despite remarkable progress in our understanding of carcinogenic process, the mechanisms of action of most chemopreventive agents have not been fully elucidated. Their general mechanism of action is shown in Fig. 20.2 along with mechanism of topoisomerase II poisons. There are many examples of therapeutic drugs which are potent catalytic inhibitors of topoisomerase II (Andoh and Ishida 1998) such as dexrazoxane (Hasinoff et al. 2001), merbarone (Pastor et al. 2012), novobiocin (Thuy et al. 2005), chloroquine (Cooper and Magwere 2008), and fostriecin (Fig. 20.3). Catalytic inhibitors of topoisomerase II do not induce cleavable complexes (Larsen et al. 2003). In case of cancer cells which are sensitive toward topoisomerase II catalytic inhibitors, triggering of checkpoint in DNA decatenation is considered to delay the entry of cells into mitosis during G2 phase (Downes et al. 1994).
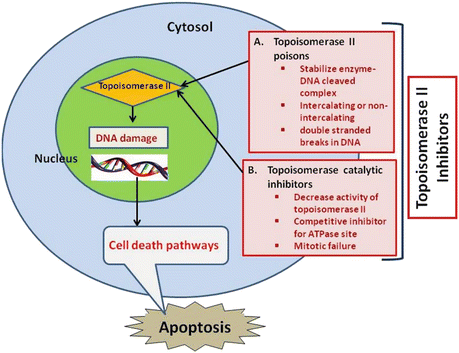
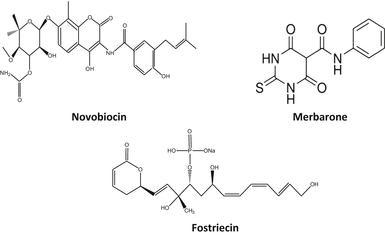
Fig. 20.2
Proposed mode of action of chemotherapeutic agents acting as topoisomerase II inhibitors in cancer cells
Fig. 20.3
Chemical structures of some topoisomerase II catalytic inhibitors
20.3.1 Merbarone
Merbarone is a catalytic inhibitor of topoisomerase II. It is a derivative of barbituric acid and chemically known as 5-(N-phenylcarboxamido)-2-thiobarbituric acid. It has exhibited activity against L1210 leukemia and some murine models. Merbarone inhibits catalytic activity of topoisomerase II by blocking DNA cleavage by enzyme. Studies by Pastor et al. (2012) in cultured CHO cells have shown that merbarone is cytotoxic as well as genotoxic and capable of inducing endoreduplication. Merbarone is known to compete with etoposide implying that both inhibitors have similar binding sites on topoisomerase II. In living cells merbarone leads to G2 arrest, S phase retardation, and formation of DNA double-strand breaks. To investigate the possible mechanisms, Wang et al. (2007) compared the cell cycle specificity of clastogenic effects of merbarone in V79 cells and demonstrated that it can cause a prolonged S phase followed by G2 delay. Merbarone can induce the release of high molecular weight DNA fragments from nuclear matrix of HL-60 leukemia cells, which precedes inter-nucleosomal size DNA fragmentation characteristic of late-stage apoptosis. It is also known to induce rapid activation of mitochondrial apoptotic pathway (Otake et al. 2006).
20.3.2 Fostriecin
Its chemical name is (6R)-5,6-Dihydro-6-[(1E,3R,4R,6R,7Z,9Z,11E)-3,6,13-trihydroxy-3-methyl-4-(phosphonooxy)-1,7,9,11-tridecatetraenyl]-2H-pyran-2-one sodium salt. It is an antitumor antibiotic obtained from Streptomyces pulveraceus. Walsh et al. (1997) have shown that it has inhibitory activity against serine/threonine protein phosphatase types 1 and 2A. It is active in vivo (Hastie and Cohen 1998). It is active against different types of leukemia in mice but less active against solid tumors due to transport deficiencies (Jackson et al. 1985; Leopold et al. 1984). fostriecin is reported to inhibit the catalytic activity of partially purified type II topoisomerase from Ehrlich ascites carcinoma with an IC50 of 40 μM (Boritzki et al. 1988).
20.3.3 Novobiocin
Also known as albamycin or cathomycin, is an aminocoumarin antibiotic produced by Streptomyces niveus. Its IUPAC name is “4-Hydroxy-3-[4-hydroxy-3-(3-methylbut-2-enyl)benzamido]-8-methylcoumarin-7-yl-3-O-carbamoyl-5,5-di-C-methyl-α-l-lyxofuranoside.” It has a half-life of 6 h. Novobiocin acts as competitive inhibitor for ATPase site of topoisomerase II critical for enzyme–DNA dissociation which can be related to its cytotoxic property against cancer cells, although some associated toxic effects on normal cells are also reported such as impairment of ATP metabolism in mitochondria and RNA polymerase inhibition.
20.4 Topoisomerase II Poisons
Chemicals that increase the level of topoisomerase II-DNA-cleaved complexes are called topoisomerase II poisons. These are further subdivided into intercalating and non-intercalating poisons. The intercalators include doxorubicin and other anthracyclines such as mitoxantrone, 4′[(9-acridinyl)amino]-methanesulphon-m-anisidide (m-AMSA), and aclarubicin (Koceva et al. 2005), and a range of other compounds that are not currently in clinical use, e.g., amonafide and ellipticine (Fig. 20.4). Non-intercalating poisons include epipodophyllotoxins such as etoposide, teniposide, and fluoroquinolones which are mainly active against eukaryotic topoisomerase II. As non-intercalating poisons do not interact strongly with DNA, it has been suggested that protein–drug interactions have key roles in the ability of topoisomerase II poisons to trap topoisomerase II covalent complexes (Nitiss 2009b). Some examples are adriamycin and etoposide (Andoh and Ishida 1998). More efficacious and novel anticancer drugs are being developed due to advancements and novel insights into physiological functions of topoisomerase II. Novel topoisomerase II inhibitors are exemplified by merbarone, aclarubicin, suramin, novobiocin, fostriecin, and quinoline derivatives (Thakur 2011).
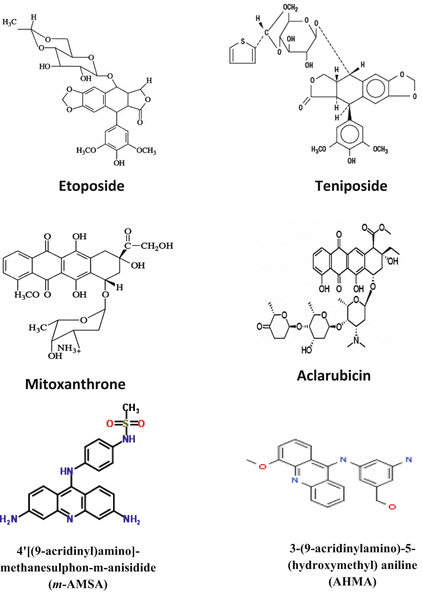
Fig. 20.4
Chemical structures of some topoisomerase II poisons
20.4.1 Etoposide
Source
Derivative of podophyllotoxin, a toxin found in Podophyllum peltatum.
Drug description
It is given in the form of etoposide phosphate which is a water-soluble ester of etoposide. Its molecular weight is 588.56 and molecular formula C29H32O13.
Pharmacology and mechanism of action
The metabolic studies of etoposide show the formation of metabolites in lactone and dimethoxyphenol rings in its structure (Van Maanen et al. 1988). The oxidative transformation of its dimethoxyphenol ring leads to products that can cause DNA damage and may play a role in drug’s mechanism of action. The capacity of drug to interfere with scission-reunion reaction of topoisomerase II by stabilizing a cleavable complex explains DNA lesions. Etoposide stabilizes the complex formed by topoisomerase II and 5′-cleaved ends of DNA thereby forming stable, non-repairable protein-linked DNA double-strand breaks in cancer cells. Cells are then eliminated by apoptosis (Karpinich et al. 2002). Etoposide is cell cycle phase specific with predominant activity occurring in late S and G2 phases.
Clinical uses
Used in treatment of some neoplasms including small cell lung cancer, Kaposi’s sarcoma, testicular cancer, acute leukemia, and lymphoma. Its current therapeutic use is limited by myelosuppression, particularly neutropenia (Kobayashi and Ratain 1994).
Toxicity and side effects
Low blood pressure, hair loss, pain and/or burning at intravenous site, constipation, and bone marrow suppression.
Interactions
Almost 30 drugs are known to interact with etoposide such as leflunomide, deferiprone, influenza virus vaccine, mumps virus vaccine, and small pox vaccine.
20.4.2 Teniposide
Source
Commonly known as VM-26. It is a semisynthetic derivative of podophyllotoxin obtained from Podophyllum peltatum.
Drug description
A white to off-white crystalline powder with molecular weight 656.66. Teniposide is insoluble in water and ether. It is slightly soluble in methanol and highly soluble in acetone and dimethylformamide.
Pharmacology and mechanism of action
It prevents tumor cells from entering mitosis and acts in late S or early G2 phase. Its mechanism of action is related with inhibition of topoisomerase II by acting as a non-intercalating poison. Cytotoxic effects of teniposide are related to a relative number of double-stranded DNA breaks produced in cells which are a reflection of stabilization of topoisomerase II-DNA intermediate (Stahelin and von Wartburg 1991; Relling et al. 2000). Topoisomerase IIα appears to be the isoform that is preferentially linked to newly replicated DNA in cells treated with teniposide.
Clinical uses
Used in treatment of childhood acute lymphocytic leukemia. Teniposide exhibits activity in glioblastoma, in neuroblastoma and in brain metastases secondary to small cell lung carcinoma (Ettinger et al. 2002).
Toxicity and side effects
Leads to severe myelosuppression, sepsis, nausea, and vomiting. Transient hypotension is also observed following rapid intravenous administration, hypersensitivity reactions characterized by chills, tachycardia, bronchospasm, dyspnea, blood pressure changes, acute central nervous system depression, and alopecia (Felix 2001).
Interactions
Therapeutically relevant concentrations of tolbutamide, sodium salicylate, and sulfamethizole are known to displace protein-bound teniposide in fresh human serum.
20.4.3 Mitoxantrone
Source
A substituted aglyconic anthraquinone derived from quinizarin found in roots of Rubia tinctorum. Quinizarin is an orange or red-brown crystalline powder which is derived from anthraquinone by replacement of two hydrogen atoms by hydroxyl groups.
Drug description
Its trade name is Novantrone and half-life is 6–12 min. Steady-state volume of distribution in tissues exceeds 1,000/m2.
Pharmacology and mechanism of action
Mitoxantrone is a substituted anthraquinone and has features known to be essential for DNA intercalation. However, Alberts et al. (1985) have found that mitoxantrone binds DNA in intact L1210 leukemia cells by non-intercalative, electrostatic interaction and induces both protein-associated and non-protein-associated DNA strand scissions. A possible mechanism for therapeutic efficacy of mitoxantrone in treatment of multiple sclerosis is that it may inhibit migration of inflammatory cells into and within the central nervous system. DNA damage resulting due to poisoning of topoisomerase II by mitoxantrone acts as a signal for NF-kappa B activation and induction of apoptosis (Boland et al. 2000). Mitoxantrone is 78 % bound to plasma proteins (Scott and Figgitt 2004; Wiseman and Spencer 1997).
Clinical uses
Used to treat acute myeloid leukemia, non-Hodgkin lymphoma, and advanced breast and advanced primary liver cancer.
Toxicity and side effects
Congestive heart failure may occur months or years after termination of therapy. Considerable evidence exists to suggest that mitoxantrone undergoes extensive metabolism, probably in liver. Abnormal liver function leads to decreased rates of total body mitoxantrone. Most important route of mitoxantrone elimination appears to be fecal (Alberts et al. 1985).
Interactions
Almost 32 drugs are known to have major interactions with mitoxantrone some of which are anzemet, teriflunomide, certolizumab, clozapine, deferiprone, fingolimod, thalidomide, etc.
20.4.4 Anthracycline Antibiotics
Anthracyclines are a class of antineoplastic drugs derived from Streptomyces sp. First anthracyclines were isolated from Streptomyces peucetius and were named doxorubicin and daunorubicin. Anthracyclines have played an important role in treatment of breast and ovarian cancer (Vejpongsa and Yeh 2014). However, toxicity of these chemicals has limited their clinical applications. They are known to cause irreversible myocardial damage (Zhang et al. 2012) which is due to build up of free radicals or metabolic products in the heart. Cardiotoxicity may also be caused due to inhibition of topoisomerase IIβ in cardiomycetes or interference with ryanodine receptors of sarcoplasmic reticulum (Fogli et al. 2004; Park et al. 2005).
Related posts:
 Prevalence of Gastrointestinal Cancers in India
Prevalence of Gastrointestinal Cancers in India
 Role of Osteopontin in Tumor Microenvironment: A New Paradigm in Cancer Therapy
Role of Osteopontin in Tumor Microenvironment: A New Paradigm in Cancer Therapy
 TG2: Player That Dictates the Rules in Cancer Progression
TG2: Player That Dictates the Rules in Cancer Progression
 Seizing Cancer Completely Through Specific Ablating Cancer Stem Cell: The Royal Road to Chemoquiescence
Seizing Cancer Completely Through Specific Ablating Cancer Stem Cell: The Royal Road to Chemoquiescence
 Current Treatment for Gallbladder Cancer
Current Treatment for Gallbladder Cancer
 Role of Surfactants in Regulation of Cancer Growth
Role of Surfactants in Regulation of Cancer Growth
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


