Fig. 2.1
Synthetic lethality. (a) Idealized DNA repair pathways in a normal cell that process a specific DNA lesion, with primary and secondary pathways catalyzed by enzymes 1–3 or a–c, respectively. (b) Cancer cell lacking enzyme 1 shifts repair from the primary to secondary pathway. Inhibition (or mutation) of any step in the secondary pathway is synthetically lethal. (c) Activation of tertiary pathway rescues the cell from synthetic lethality, producing a cancer cell resistant to the inhibitor. The tertiary pathway may be error-prone; the cell may survive the damage but with increased mutation load (genome instability) due to misrepair, which can drive tumor progression and further resistance to subsequent therapy
2.2 DNA Repair Mechanisms
2.2.1 Base Excision Repair, Nucleotide Excision Repair, and Mismatch Repair
There are five classes of DNA repair pathways, each with two or more subtypes (Fig. 2.2). Three sets of pathways act on damage present on single strands. Base excision repair (BER) comprises several subpathways that repair non-bulky DNA lesions such as ring-opened bases and small adducts like oxidized bases (Fig. 2.2a). The first step in BER is lesion recognition by one of several glycosylases that remove the damaged base, producing an abasic site. PARP1, APE1 endonuclease, and deoxyribophosphodiesterase activities create a single-strand break and DNA repair is completed by DNA polymerase and DNA ligase activities (Krokan and Bjoras 2013). Nucleotide excision repair (NER) processes bulky lesions that distort the double helix, including pyrimidine dimers and large DNA adducts Kamileri et al. 2012) (Fig. 2.2b). NER requires lesion recognition proteins and endonucleases that create single-strand breaks ~15 nt from the lesion, a helicase removes the ~30 nt oligonucleotide carrying the lesion, DNA polymerase then fills the single-strand gap and ligase completes the repair. Mismatch repair (MMR) differs from all other DNA repair pathways in that there is no “damage” per se, but instead chemically intact mismatched bases occur on otherwise complementary strands (Hsieh and Yamane 2008). Mismatches arise via replication errors, strand exchange during homologous recombination (HR), and deamination of 5′-methyl cytosine which produces thyimidine and a G-T mismatch. MMR also processes single- and multi-base loops that arise when bases are inserted or deleted – insertion/deletion loop mismatches can arise by replication errors (especially in sequences with mononucleotide repeats, or certain trinucleotide repeats that are prone to self-annealing as these form relatively stable hairpin structures), or during homologous recombination (see below). MMR involves mismatch recognition, single-strand nicking 5′ and/or 3′ of the mismatch from which long-tract single-strand excision removes the mismatched base(s), re-synthesis to fill the single-strand gap, and ligation (Fig. 2.2c). Long-tract MMR operates on mismatches that arise during replication and HR; G-T mismatches can be repaired by long-tract MMR or by a specialized G-T MMR system that is more akin to BER as it is initiated by a G-T specific glycosylase (Bill et al. 1998; Wiebauer and Jiricny 1990). BER, NER, and MMR are relatively accurate repair mechanisms because each uses an intact complementary strand opposite the lesion to direct repair. However, repair polymerases tend to be less accurate than replicative polymerases, and this can result in localized mutagenesis. In addition, BER proceeds through abasic intermediates that can be subject to translesion DNA synthesis by Y-family polymerases, which are low processivity, error-prone DNA polymerases, providing another path to localized mutagenesis (Simonelli et al. 2005). There are also examples where the accuracy of DNA repair systems are downregulated to specifically enhance mutagenesis, e.g., MMR induced trinucleotide repeat expansion and antibody maturation (Pena-Diaz and Jiricny 2012).
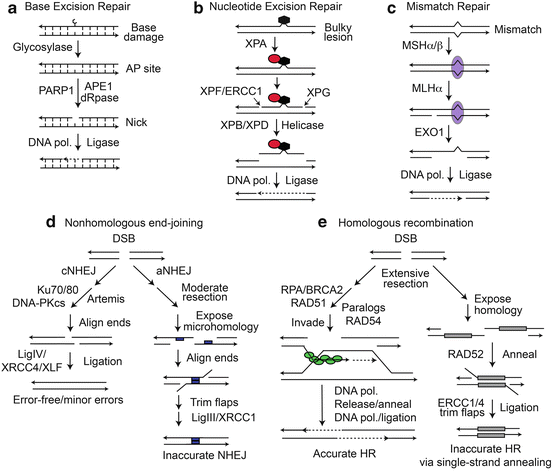
Fig. 2.2
DNA repair pathways. (a) Repair of base damage by BER results in short patch repair and is promoted by PARP1. (b) Bulky lesion repair by NER removes a ~30 nt single strand oligonucleotide carrying the lesion. (c) MMR involves long-patch excision and resynthesis initiated at nicks distant from the mismatch. (d) NHEJ includes relatively accurate cNHEJ, and inaccurate aNHEJ pathways distinguished by the extent of end resection, requirement for microhomology (blue boxes). (e) HR catalyzed by RAD51 (green ovals) is generally accurate. BRCA1 and FANC proteins (not shown) also function in RAD51-dependent HR. SSA between linked repeats (grey boxes) deletes one repeat and intervening sequences
2.2.2 Features and Roles of DSBs and DSB Repair Pathways
DSBs are the most important DNA lesion because they can trigger genome rearrangements and unrepaired DSBs are usually lethal. DSBs are induced by ionizing radiation, and by endogenous nucleases during meiosis (SPO11) and V(D)J recombination (RAG1/2) (Keeney and Neale 2006; Nishana and Raghavan 2012). AID deaminates cytosine to uracil which can be processed to staggered single-strand breaks to create DSBs that trigger immunoglobulin class switch recombination or gene conversion (Daniel and Nussenzweig 2013), which along with V(D)J recombination are important mechanisms for generating antibody diversity. DSBs also arise during DNA replication when forks encounter blocking lesions (single-strand breaks, many types of base damage, most DNA adducts, pyrimidine dimers, and intra- and inter-strand crosslinks) (Allen et al. 2011; Budzowska and Kanaar 2009). DSBs are marked by phosphorylated histone H2AX (γ-H2AX) (Ward and Chen 2001) which plays important roles in DNA damage checkpoint signaling and DSB repair (Chanoux et al. 2008; Downey and Durocher 2006).
DSBs are repaired by nonhomologous end-joining (NHEJ) and HR (Fig. 2.2d, e). DSB repair by NHEJ is frequently inaccurate, yielding short (1–20 nt) deletion or insertion mutations (Deriano and Roth 2013). When two DSBs occur simultaneously on different chromosomes, NHEJ can mediate translocations (Lieber et al. 2006; Nickoloff et al. 2008; Weinstock et al. 2006). HR is generally accurate, but since HR can occur between any two homologous sequences (sister chromatids, homologous chromosomes, linked repeats in inverted or direct orientation, and repeats on non-homologous chromosomes), HR poses significant risks of medium- to large-scale genome rearrangements including deletions, inversions, amplifications, small- to large-scale loss of heterozygosity (LOH), and translocations (Nickoloff 2002).
2.2.3 DSB Repair by Nonhomologous End-Joining
NHEJ comprises two pathways, classical and alternative NHEJ (cNHEJ, aNHEJ) (Fig. 2.2d). Although both pathways are inaccurate, cNHEJ is more accurate and is the dominant DSB repair pathway in mammalian cells; aNHEJ appears to serve as a backup pathway to cNHEJ as it is typically observed when cells have a defect in a cNHEJ factor (Deriano and Roth 2013; Weinstock et al. 2007; Wray et al. 2010, 2013). cNHEJ involves little to no end-resection, whereas moderate end-resection is key to exposing microhomologies on complementary strands central to the aNHEJ pathway (aNHEJ is sometimes called “microhomology-mediated end-joining”). 53BP1 and BRCA1 are implicated in regulation of end-resection, which in turn regulates cNHEJ – aNHEJ pathway choice (and NHEJ – HR choice; see below) (Deriano and Roth 2013; Panier and Boulton 2014; Symington and Gautier 2011).
cNHEJ initiates with Ku70/Ku80 binding to DSB ends to which DNA-PKcs is recruited, activating its kinase. Artemis is a nuclease required for processing certain types of broken ends (Jacobs et al. 2010). Metnase is a recently evolved nuclease and protein methylase that promotes cNHEJ by enhancing recruitment and retention of other NHEJ factors by methylating histone H3 (Fnu et al. 2011), and possibly through nucleolytic end processing (Hromas et al. 2008). Together these factors promote association of broken ends with little to no base-pairing (Lieber 2010). Prior to ligation by Ligase IV and accessory factors XRCC4 and XLF, DNA-PKcs phosphorylation by itself or ATM stimulates its dissociation from ends (Dahm 2008; Lieber 2010). Genetic defects in, or inhibition of, cNHEJ factors shunts DSBs to aNHEJ, which depends on moderate end-resection by MRE11-RAD50-NBS1 (MRN) and CtIP to expose microhomologies, and requires PARP1 and Ligase III-XRCC1 (Deriano and Roth 2013; Rupnik et al. 2010; Wray et al. 2013; You et al. 2009). By suppressing resection by MRN and CtIP, 53BP1 prevents aNHEJ and thus reduces the risk of aNHEJ-mediated translocations (Bothmer et al. 2010). PARP1 inhibitors also prevent aNHEJ-mediated translocations, and may be useful in reducing the risk of oncogenic translocations associated with cancer chemotherapy (Wray et al. 2013).
2.2.4 DSB Repair by Homologous Recombination
HR comprises two pathways, a conservative, accurate pathway that involves strand invasion mediated by RAD51, and a non-conservative, error-prone, RAD51-independent pathway termed single-strand annealing (SSA) (Fig. 2.2e). Both HR pathways require extensive end-resection (100–1,000s of bases). Cells with defects in RAD51-dependent HR shunt DSBs to SSA, destabilizing the genome (Tutt et al. 2001). DSB repair pathway choice is apparently regulated by proteins that control resection, including 53BP1 and BRCA1 (Panier and Boulton 2014), with increasing resection along a cNHEJ – aNHEJ – HR continuum. The extensive resection required for HR begins with end-processing by MRN-CtIP followed by BLM helicase and two nucleases, DNA2 and EXO1 (Symington and Gautier 2011). RAD51-dependent HR can lead to localized LOH termed gene conversion, a mechanism that nearly always results in unidirectional (non-reciprocal) transfer of information from an unbroken donor molecule to a broken homologous molecule during DSB repair – the LOH region is termed a gene conversion tract and these can range from just a few bp to many kbp. Gene conversions are sometimes associated with reciprocal exchange of sequences flanking the conversion tract, and during HR between homologous chromosomes, 50 % of crossovers can cause LOH of an entire chromosome arm, extending from the point of the crossover to the telomere (Nickoloff 2002). In cells with defects in HR proteins, RAD51-mediated strand exchange may abort after the strand invasion and repair synthesis is initiated but before second end capture. In this case, synthesis continues to the end of the donor chromosome, which also results in LOH from the DSB to the telomere in a process termed “break-induced replication” (Llorente et al. 2008). As noted above, crossovers also pose significant risk of large-scale genome rearrangement, and are suppressed by proteins like BLM (Cheok et al. 2005). RAD51-dependent HR is generally restricted to S and G2 phases when sister chromatids serve as closely associated, essentially 100 % accurate repair templates with low risk of large-scale genome rearrangement.
End resection produces long, 3′ ssDNA tails bound by RPA that is exchanged for RAD51 in a reaction promoted by “mediator” proteins BRCA2, RAD52, and RAD51 paralogs (XRCC2, XRCC3, RAD51B, RAD51C, and RAD51D). The RAD51-ssDNA nucleoprotein filament searches for and invades homologous sequences, the invading strand is extended by DNA polymerase, and then released and “captured” by the resected end on the opposite side of the DSB. This one-ended invasion mechanism, termed synthesis-dependent strand annealing (SDSA), poses low risk of crossovers (Fig. 2.2e). In some cases, both ends invade, creating a double Holiday junction intermediate that is resolved by BLM-TOP3α-RMI1-2 without crossing over (Heyer et al. 2010). In the absence of BLM, crossovers are much more frequent and this confers a genome-instability and cancer-prone phenotype (Cheok et al. 2005). Some RAD51 mediator proteins also function later in HR, stabilizing the strand invasion intermediate and removing RAD51 from ssDNA after the RAD51-ssDNA filament invades the homologous sequence to prepare for repair synthesis (Brenneman et al. 2002; Fortin and Symington 2002). RAD54 (and possibly RAD54B) also act late, altering chromatin in donor sequences to promote strand invasion (Heyer et al. 2010).
The SSA pathway (Fig. 2.2e) is RAD51-independent and at least in yeast, depends on the strand annealing activity of RAD52 (Heyer et al. 2010). SSA involves extensive resection to expose complementary sequences in linked, direct repeats which anneal to produce a deletion product lacking one of the repeats and the intervening sequences. SSA can also mediate translocations (Weinstock et al. 2006). SSA and RAD51-mediated gene conversion are competing HR pathways, and the balance shifts toward SSA for closely spaced repeats (Schildkraut et al. 2005).
2.2.5 Role of HR in Replication Stress Responses
Nearly all DNA lesions, whether induced by chemotherapeutics or ionizing radiation, block replicative DNA polymerases, causing “replication stress.” RAD51-mediated HR plays a critical role in restarting stalled and collapsed replication forks, and it is this role that accounts for the fact that RAD51 is an essential protein in mammalian cells (Allen et al. 2011). When replication forks encounter blocking lesions, the fork stalls and the replisome is stabilized by DNA repair and checkpoint proteins including RPA, ATR-ATRIP, ATM, BLM, and INO80 (Budzowska and Kanaar 2009; Davies et al. 2007; Shimada et al. 2008; Zou et al. 2006). If a stalled fork is not restarted in timely manner, it collapses to a DSB. Unlike DSBs induced by nucleases or ionizing radiation, fork collapse yields single-ended DSBs that cannot be readily repaired by NHEJ. Like two-ended DSBs, one-ended DSBs are marked by γ-H2AX, and the induction and resolution of this signal are measures of fork collapse and repair (De Haro et al. 2010; Kim et al. 2014). Stalled and collapsed replication forks can be restarted by several HR-related mechanisms (Allen et al. 2011; Budzowska and Kanaar 2009); one mechanism is shown in Fig. 2.3. A fork stalled by a blocking lesion can “regress”, and this allows the lesion to be bypassed via an HR mechanism (Budzowska and Kanaar 2009). Lesion bypass via fork regression is not a repair mechanism, but a damage tolerance mechanism. Another type of damage tolerance mechanism is mediated by error-prone, translesion synthesis (TLS) polymerases (Sale 2013). Understanding tumor cell responses to replication stress, and in particular, the role of checkpoint and HR pathways in these responses, has emerged as a critical topic in cancer biology.
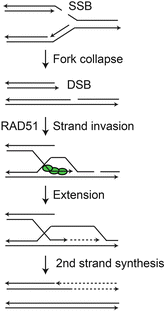
Fig. 2.3
Restart of collapsed replication fork. Fork encounter with lesion (single-strand break in this example) causes collapse to one-ended DSB. RAD51 loads onto resected 3′ end that invades the sister chromatid, is extended, and then serves as template for second strand synthesis to complete DNA replication
2.3 DNA Repair Pathway Regulation and Networks
DNA repair pathways are highly regulated and exist in complex, interacting networks. DNA repair can be regulated in many ways. For example, repair proteins may be absent (gene knockout) or exist as hypomorphs with reduced or altered function; mRNA levels can be altered by transcription factors, microRNAs, and other factors that regulate mRNA stability; and protein stability and function can be altered by posttranslational modifications such as phosphorylation, ubiquitylation, and SUMOylation. There is evidence that oxidative damage upregulates both BER and NER (Cabelof et al. 2002; Kirkali et al. 2011), and gene expression profiling has shown that DNA damage upregulates many DNA repair genes (Friedberg et al. 2005). At a functional level, exposure of cells to low levels of DNA damage confers resistance to a subsequent higher dose, so-called adaptive responses (Huang et al. 2006; Preston 2005). Adaptive responses are protective, which is beneficial for normal tissue, but can have the negative effect of conferring resistance of the tumor to treatment. Adaptive responses may reflect broad effects of damage on repair, checkpoint, and programmed cell death pathways, rather than upregulation of specific DNA repair pathways. For example, there is no clear evidence for upregulation of HR by DSB damage (Heyer et al. 2010), yet adaptive responses to ionizing radiation have been described (Preston 2005).
As noted above, DSB repair pathway choice is regulated by resection of DSB ends in a cell cycle dependent manner (Durant and Nickoloff 2005; Shrivastav et al. 2008; Symington and Gautier 2011). The upregulation of HR during S and G2 phase probably reflects the combined effects of end resection and availability of sister chromatid repair templates. When considering synthetic lethal or sensitivity approaches to cancer therapy, DSB repair illustrates an important principal: a single type of lesion can be shunted to multiple repair pathways that operate in hierarchical fashion (Fig. 2.1). In the case of DSBs, this hierarchy runs from cNHEJ to aNHEJ to HR. Moreover, DNA repair pathways display considerable functional overlap. As just one example of network connectivity, NER has known functional interactions with MMR, BER, HR, NHEJ, and TLS (Shaheen et al. 2011). There are several types of interactions among repair pathways, including shared repair factors, processing of specific lesions by multiple pathways, and common repair intermediates produced by different pathways.
DNA repair pathways are also tightly integrated with DNA damage checkpoint pathways. DNA damage checkpoints were originally defined as damage sensor, signaling and effector pathways that arrested cells in specific phases of the cell cycle, ostensibly to allow time for repair before resuming the cell cycle to reduce problems associated with replication or segregation of damaged DNA. However, studies in yeast demonstrated that artificially arresting certain checkpoint-defective mutants failed to rescue damage sensitivity (DeMase et al. 2005; Redon et al. 2003; Toh et al. 2006). There is now clear evidence that checkpoint factors, such as ATM and γ-H2AX regulate both checkpoints and DNA repair (Downey and Durocher 2006; Shrivastav et al. 2009; Smith et al. 2010; Xie et al. 2004).
The tumor microenvironment imposes considerable stress on tumor cells, including glucose and oxygen deprivation, low pH, replication stress associated with activated oncogenes, and of course, there is stress induced by genotoxic therapeutics (Bartkova et al. 2006; Gozuacik and Kimchi 2004; Karantza-Wadsworth et al. 2007; Mathew et al. 2007). There is substantial evidence that cells actively upregulate mutagenesis in response to stress by at least two mechanisms: DSB-induced gene amplification, which can increase expression of proteins that confer stress resistance, and switching from accurate to error-prone DSB repair mechanisms. These processes underlie “adaptive mutagenesis” which helps generate mutants that are better adapted to a stressful environment, and in essence, reflect “regulated evolvability” (Galhardo et al. 2007; Gonzalez et al. 2008; Hastings et al. 2009; Ponder et al. 2005). The inherent complexity of DNA repair networks and their regulation, coupled with the immense genetic heterogeneity of solid tumors, presents both challenges and opportunities for developing synthetic lethal and sensitivity approaches to cancer treatment.
2.4 DNA Repair and Genome Instability: Roles in Cancer Etiology, Tumor Progression and Resistance to Therapy
Genome instability was recognized early as a hallmark of cancer. Cancer arises when cells acquire altered cell growth properties including independence from growth signals, immortality, and defects in programmed cell death pathways. Tumor progression to a more aggressive state depends on alterations that affect tissue responses including angiogenesis, tissue invasion, and adaptability to new tissue environments which drive metastasis. Although it has long been known that cancer cells have unstable genomes, it was difficult to establish whether genome instability was an early driver of cancer, or a secondary manifestation of the cancer state. There are now several well-established cases where genome instability has been definitively shown to precede cancer (Hanks et al. 2004; Lengauer et al. 1998; Pikor et al. 2013; Shih et al. 2001; Weaver et al. 2007). Although the number and types of genetic changes required to initiate tumorigenesis and promote tumor progression vary among different types of cancers, a common theme is the early acquisition of defects in DNA repair systems that play critical roles in genome stabilization.
Genome instability manifests over a wide scale, from point mutations, trinucleotide repeat expansions and contractions, gene duplications, deletions, and inversions, to large-scale chromosome changes including translocations and whole chromosome gains and losses. Defects in DNA repair pathways contribute to specific instabilities (Fig. 2.4). Point mutagenesis is greatly increased by defects in BER, NER, and MMR. MMR also suppresses microsatellite repeat expansion or contraction reflecting replication slippage at short repeats such as trinucleotide repeats. cNHEJ and RAD51-dependent HR suppress translocations by aNHEJ and SSA, respectively.
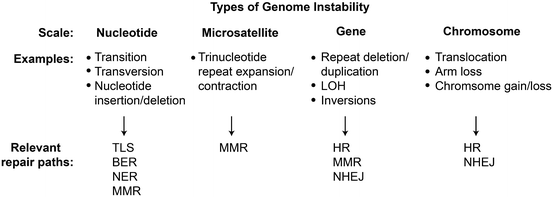
Fig. 2.4
Genome instability and DNA repair. Genome instabilities result in small, moderate, and large scale genome alterations. Indicated repair pathways suppress or induce different types of instabilities
Thus, DNA repair defects cause genome instability, which accelerates the acquisition of mutations in critical growth regulatory genes, including gain-of-function mutations in proto-oncogenes and loss-of-function mutations in tumor suppressor genes (Wang 2005). Defects in specific DNA repair pathways are associated with heritable cancer syndromes such as colon cancer (MMR), skin cancer (NER), and breast cancer (HR) (Boulton 2006; Friedberg 2001; Gudmundsdottir and Ashworth 2006; Jass 2002; Jiricny 2006; Venkitaraman 2002).
Conversion of a normal cell to a metastatic cancer cell may require 3–10 mutations in key cell growth and tissue regulatory genes (Ilyas et al. 1999; Schedin and Elias 2004; Spurgers et al. 2006). Mutation rates in normal mammalian cells are low, ~10−10 per base per cell generation, which translates to 10−6 to 10−8 mutations per gene per cell generation (Baer et al. 2007; Roach et al. 2010). Assuming mutations arise independently, even at the higher rate of 10−6 mutations per gene per generation, the odds of accumulating as few as five critical mutations in a single cell is vanishing small (~10−32). DNA repair defects greatly increase mutation rates and the odds of accumulating critical mutations.
Mutations arise infrequently in undamaged DNA, e.g., by base mis-incorporation during DNA replication, but mutation rates are dramatically increased at or near sites of DNA damage as a result of inaccurate DNA repair or error-prone lesion bypass, including translesion DNA synthesis and recombinational mechanisms (Nickoloff 2002; Sale 2013; Shaheen et al. 2011). DNA lesions can arise spontaneously, reflecting the chemical lability of DNA (e.g., deamination of cytosine and 5-methyl cytosine to uracil and thymidine, respectively); damage caused by reactive oxygen species (ROS) formed during normal cell metabolism; and single- and double-strand breaks created by nucleases or when replication forks collapse (Allen et al. 2011; Barnes and Lindahl 2004; Caldecott 2008; Gates 2009). DNA oxidation by ROS is a major source of mutations as it produces many types of lesions including oxidized bases (e.g., 8-oxoguanine), abasic sites, bulky lesions (e.g., etheno and protein-DNA adducts), strand breaks, and DNA crosslinks (Waris and Ahsan 2006). Genotoxic agents are mutagenic because DNA repair does not always accurately restore the original DNA sequence.
When cells acquire a DNA repair defect, increased mutagenesis drives early stage tumorigenesis through alterations in key growth regulatory genes, and it also drives the rapid evolution of tumor cells that promotes tumor progression. As long as mutation rates remain below a critical “error catastrophe” limit (Fox and Loeb 2010), high mutation rates allow tumor cells to “test” mutations that allow them to adapt to various types of stress, including nutrient and oxygen deprivation, a common feature of tumor microenvironments, and to develop resistance to therapy. Most cancer patients are treated with DNA damaging agents, even when tumors are resectable, including chemotherapeutic drugs and/or ionizing radiation. These approaches exploit the fact that tumor cells divide more rapidly than normal cells, and cells actively replicating DNA are highly susceptible to the cytotoxic effects of DNA damage. This is because nearly all types of DNA damage block replicative DNA polymerases, causing forks to stall and eventually collapse to cell-lethal DSBs (Allen et al. 2011; Branzei and Foiani 2010). Nonetheless, tumor cells can be highly resistant to traditional therapies. It seems paradoxical that tumorigenesis can be driven by defects in DNA repair genes yet the resulting rapidly growing cells are often resistant to DNA damaging agents. Although in certain cases this may reflect the use of genotoxins that create specific types of DNA damage that are repaired by pathways that remain functional in a particular tumor (i.e., failure to appropriately tailor the treatment to the specific DNA repair defect), there are many other ways to resolve this paradox. For example, resistance to therapy may reflect upregulation of other DNA repair pathways or drug efflux pathways, and/or defects in apoptosis and other programmed cell death pathways that are normally triggered by heavy loads of DNA damage. In this light it is noteworthy that ~50 % of tumor cells carry defects in p53, which plays critical roles in apoptosis (Carvajal and Manfredi 2013).
As noted above, the increase in genome instability associated with DNA repair defects promotes the acquisition of mutations, including those that drive changes in other DNA repair, drug efflux, and programmed cell death pathways. The realization that rapid evolution of tumor cells allows them to adapt to stressful environments and acquire resistance to genotoxic therapeutics, has forced cancer biologists to reevaluate therapeutic strategies. The traditional approach to induce DNA damage in tumor cells with chemo- and/or radiotherapy is fairly effective at killing bulk tumor cells, but this damage can also generate (and ultimately select for) a subpopulation of tumor cells that are resistant to therapy, and moreover, potentially generate more aggressive tumor cells leading to local tumor recurrence and progression to a more dangerous, metastatic state. These traditional approaches were initially promising because they provide clear short-term benefits, namely rapid and marked reduction in bulk tumor mass and increased median survival times, but they do not necessarily increase long-term patient survival (Fig. 2.5a). To increase long-term patient survival, i.e., to increase the “tail” of Kaplan-Meier survival curves (Fig. 2.5b), we must shift our focus toward strategies that kill or prevent proliferation of essentially 100 % of tumor cells. The significant difference between median survival time and the fraction of long-term survivors was elegantly explained by Stephen Jay Gould (1985). While the goal of improving long-term survival is clear, achieving this goal presents major challenges given the difficulty in eradicating tumor cells while minimizing the effects of chemo- or radiotherapy on normal tissue. In addition to causing serious immediate side effects such as neurological and gastrointestinal problems, fatigue, fever, liver and kidney failure, traditional cancer therapies can cause a wide range of serious late effects including cardiac disease, nephrotoxicity, infertility, hearing loss, neurological problems, and secondary tumors (Gururangan 2009). These late effects are a more serious problem for pediatric patients and others with potential for long-term survival. Therapeutic interventions that target tumor weaknesses through synthetic lethal and synthetic sensitivity approaches that exploit known DNA repair, checkpoint, or programmed cell death defects (or target these pathways with inhibitors) could be more effective at eradicating tumor cells while minimizing harm to normal tissues. Because DNA repair plays such a prominent role in tumorigenesis and tumor response to therapy, a deep understanding of DNA repair networks holds significant promise for improving cancer therapy.
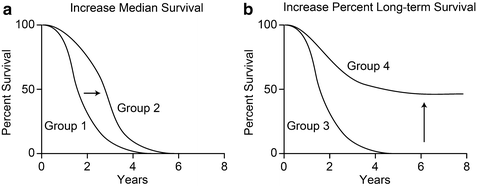
Fig. 2.5
Idealized Kaplan-Meier survival curves showing (a) increased median survival time of treatment group 2 vs group 1, but no increase in long-term survival, and (b) increased percentage of long-term survivors in treatment group 4 vs group 3
2.5 Synthetic Lethality and Sensitivity in Targeted Cancer Therapy
The key to effective cancer treatment is to target tumor cells while sparing normal tissue. Although there have been notable successes identifying tumor-specific targets, such as the brc-abl fusion protein in chronic myelogenous leukemia that can be inhibited with Imatinib (Gleevec) this approach may not be generally applicable to far more genetically heterogeneous solid tumors (Fox et al. 2009). This has led to the idea that the search for cures should focus on “disrupting the broader biological pathways that support cancer growth” (Hayden 2008). DNA damaging agents do indeed disrupt a critical biological pathway required for growth (DNA replication), but systemic chemotherapy, and even well-targeted radiotherapy, can cause significant normal tissue damage. Normal tissue tolerance limits the doses that can be delivered to tumors, and increases the chance that some tumor cells will survive. After therapy, surviving tumor cells are likely to have suffered considerable DNA damage which could drive mutagenesis and promote tumor progression upon recurrence. It is therefore imperative to develop targeting strategies that selectively kill tumor cells. Given that DNA repair defects are early drivers of many solid tumors, there is great interest in developing therapeutics that exploit these potential weaknesses based on synthetic lethality and sensitivity. Because unrepaired DSBs are generally lethal to cells, there has been significant focus on DSB repair pathways and genotoxins that directly or indirectly induce DSBs. However, we should not restrict our thinking to just these pathways and agents, as there are many pathways to death or even senescence, which achieve the same goal of preventing tumor growth and spread.
Related posts:
 Introduction to Cell Stress Responses in Cancer: The Big Picture
Introduction to Cell Stress Responses in Cancer: The Big Picture
 Pro-senescence Therapy for Cancer: Time for the Clinic
Pro-senescence Therapy for Cancer: Time for the Clinic
 Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
 p53 at the Crossroads Between Stress Response Signaling and Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
p53 at the Crossroads Between Stress Response Signaling and Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
 Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
 The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


