Insight into structural genomic rearrangements in cancer has first been obtained through the identification of cytogenetically visible changes, such as whole-arm translocations. The Philadelphia translocation (t(9;22)) was the first structural variation observed in cancer (Rowley 1973). This translocation occurs in 95 % of chronic myelogenous leukemia (CML) cases and results in the formation of an oncogenic fusion gene between BCR and the tyrosine kinase gene ABL1 (Shtivelman et al. 1985). These initial findings have triggered subsequent investigation of cytogenetic aberrations in cancer cells, culminating into the identification of hundreds of cancer gene fusions (Mitelman et al. 2007).
The emergence of array-based techniques such as comparative genomic hybridization (aCGH) and SNP-arrays (Box 1) has provided further crucial insight into the impact of chromosomal rearrangements in cancer genomes (Beroukhim et al. 2007). Pan-cancer analyses of thousands of datasets have uncovered genomic regions and genes that show significant change in copy number across different cancer types (Beroukhim et al. 2010; Zack et al. 2013), leading to the identification of novel cancer genes.
Patterns of structural rearrangements can be substantially refined by the use of next-generation sequencing technology. Paired-end sequencing strategies have been instrumental to detect both copy number changes and balanced rearrangements in the human genome simultaneously (Box 1) (Korbel et al. 2007). Application of this methodology to cancer genomes has led to the classification of somatic rearrangements across all size ranges and types at unprecedented resolution (Campbell et al. 2008) (Fig. 2). Sequencing data have shown that cancer genomes contain more somatic rearrangements than previously anticipated (Stephens et al. 2009). Also, rearrangement types may vary for different cancers, and even cancer subtypes. This is illustrated by the predominance of tandem duplications among the structural landscape of subsets of breast and ovarian cancers, whereas other subsets are mostly characterized by deletions (McBride et al. 2012). In addition, certain tumor types show a markedly increased number of structural changes (e.g., breast cancer and lung squamous cell cancer) when compared to others (kidney cancer) (Yang et al. 2013). Cancer genomes harbor an overrepresentation of complex changes, interchromosomal translocations and tandem duplications when compared to germline rearrangements (McBride et al. 2012; Campbell et al. 2010; Hillmer et al. 2011). Also, chromosomal rearrangements can arise throughout cancer development and metastasis, leading to the detection of both shared and lesion-specific rearrangements in several cancers (Campbell et al. 2010; Kloosterman et al. 2011a; Hoogstraat et al. 2014; Ding et al. 2010).
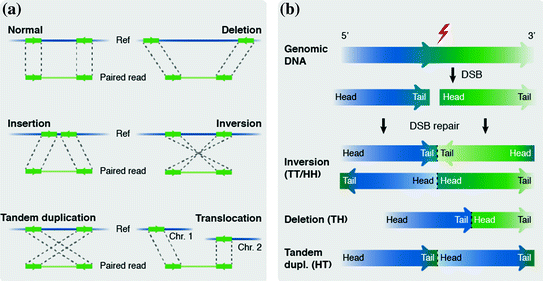
Fig. 2
The detection of genomic rearrangements from paired-sequencing data. a Individual reads are aligned to a reference genome and discordant reads indicate the presence of rearrangements (Box 1). Different SV types lead to different types of discordance; deletions show a larger distance between aligned reads compared to the original DNA fragment length, whereas insertions will be characterized by a smaller distance between aligned reads. In the case of an inversion and tandem duplication the order of the reads in the pair is swapped, for inversions the reads will map to the opposite DNA strand. In case of an inter-chromosomal translocation the reads of a read pair will map to different chromosomes. b Intrachromosomal rearrangements lead to a change in orientation of breakpoint junctions (Box 1). The orientation is indicated by using head (H), which indicates the 5′ end of a fragment (i.e. the lowest coordinate), and tail (T), indicating the 3′ end of a fragment (i.e. the highest coordinate). Different rearrangement types lead to different orientation conformations. The first letter corresponds to the fragment with the lowest coordinate and the second to the fragment with the highest coordinate. TH orientation of the breakpoint junction indicates the presence of a deletion, TT and HH indicate the presence of an inversion. HT (not TH) orientation indicates a tandem duplication, due to the fact that the first letter corresponds to the read with the lowest coordinate, which in the case of a tandem duplication is the second read in the pair
3 A Historical View on Complex Genome Rearrangements and Cancer Development
The complexity of some chromosomal aberrations in cancer was noted well before the introduction of high-resolution copy number and sequencing technologies. For example, the vast majority of leukemia’s with a Philadelphia chromosome contain a simple translocation between chromosomes 9 and 22, but very complex variants involving up to five chromosomes have also been observed (Adhvaryu et al. 1988; Kadam et al. 1990; Rosson and Reddy 1988). In addition, BCR-ABL positive cases of leukemia without a karyotypically visible rearrangement have been described (Fitzgerald and Morris 1991). Careful analysis of the BCR and ABL loci in these cases has revealed complex exchanges of chromosome segments and it was suggested that this was not a result of serial changes, but rather a single concerted event, reminiscent of chromothripsis (Fitzgerald and Morris 1991). Furthermore, cytogenetic analysis already revealed that amplification of MYCN in neuroblastoma is a process involving complex genomic rearrangements (Nishi et al. 1992). Those studies suggested that the complex and heterogeneous rearrangements of MYCN in amplicons of neuroblastoma cell lines have preceded the amplification process. The amplification of oncogenes may involve the separate propagation of chromosome fragments by formation of episomes (double-minute chromosomes). Alternatively, oncogene-containing chromosome segments can form an array within a chromosome and appear as homogeneously staining regions (HSR) (Cowell 1982; Shimizu 2009). HSRs are thought to arise via breakage-fusion bridge (BFB) cycles, a process of chromosome fusion following telomere attrition (Shimizu et al. 2005; Cowell and Miller 1983). Sequencing of amplified regions in cancer genomes revealed a wide variety of configurations, including evidence for BFB cycles and integration of double minutes into the genome (Bignell et al. 2007).
Further evidence for the importance of complex genomic aberrations for cancer development was gained through cytogenetic studies that revealed recurrently affected regions, such as the identification of complex rearrangements on 12q13 in leiomyoma (Nilbert et al. 1989). The genes in this region are now known to be important drivers for this cancer type and the underlying complex rearrangements are frequently caused by chromothripsis. Osteosarcomas have a remarkably high frequency of complex chromosomal rearrangements, many of which are recurrent (Bayani et al. 2003). Notably, osteosarcomas form a cancer type with a very high frequency of chromothripsis (Stephens et al. 2011).
Altogether, early cytogenetic studies have shown the involvement of complex genomic rearrangements in cancer gene amplification, formation of cancer fusion genes and recurrent changes on specific chromosomal regions. It is very likely that at least some of these complex rearrangements resulted from chromothripsis. The precise structure of complex rearrangements, their differences and impact on cancer genes has only become apparent based on genome sequencing analysis (Box 1). This has led to the discovery of chromothripsis as a distinct and well-discernable entity among complex chromosome rearrangements.
4 Hallmarks of Chromothripsis in Cancer Genomes
A key feature of chromothripsis is the geographic localization of rearrangements within the genome and profound clustering of breakpoints on one or multiple parts of one or a few chromosomes. The highly localized character of chromothripsis is further emphasized by the fact that all rearrangements involve a single parental chromosome (Stephens et al. 2011; Korbel and Campbell 2013). A second hallmark is the presence of frequent oscillations between two copy number states due to the loss of fragments to the cell, with loss-of-heterozygosity (LOH) in the lower copy number (deleted) regions, but retainment of heterozygosity in regions with the higher copy number (Stephens et al. 2011). Alternatively, three or more copy-number states can be observed. Chromosome fragments can be incorporated into a double-minute chromosome (Fig. 3), which can subsequently become amplified if it contains an oncogene (Stephens et al. 2011; Rausch et al. 2012). Additionally, partial chromosome duplication preceding or following chromothripsis may occur (Zhang et al. 2013). In those instances, three or more copy number states can be observed: one for lost fragments, one for retained fragments and one or more, high copy-number states for duplicated fragments and fragments of amplified double-minute chromosomes (Stephens et al. 2011; Rausch et al. 2012). As a third hallmark of chromothripsis, regions with the lower copy number are not caused by simple tail-to-head deletions, but by a series of complex breakpoint junctions that span the involved chromosomal regions. Fourth, all four possible intrachromosomal breakpoint orientations (Fig. 2b) are represented in approximately equal numbers. Finally, the overall chromosome configuration resulting from chromothripsis involves joined fragments that are often not located in the proximity of each other in the reference genome (Stephens et al. 2011).
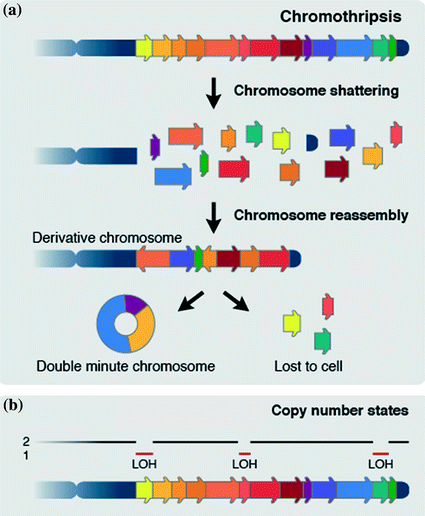
Fig. 3
Chromosome shattering and reassembly by chromothripsis. a One or multiple parts of one or a few chromosomes are shattered into pieces in a one-off catastrophic event. Chromosome are subsequently stitched back together in a random order by NHEJ, with some fragments being incorporated into a double-minute chromosome and others getting lost to the cell. b Chromothripsis typically leads to frequent oscillations between two copy number states in which the lower copy state shows loss of heterozygozity (LOH)
These hallmarks of chromothripsis have led to a model, whereby one or multiple parts of one or more chromosomes are shattered into pieces and randomly stitched back together, with some parts getting lost to the cell, and others incorporated into a double-minute chromosome (Fig. 3). This results in massively rearranged chromosomes involving tens to hundreds of rearrangements. A lower limit for the number of breaks constituting a chromothripsis event is not defined. It could be that complex rearrangements constituted by just a few or many local breakpoints are both caused by the same molecular trigger (Kloosterman and Cuppen 2013).
Chromothripsis is not restricted to cancer, but also occurs in the germline, leading to severe congenital phenotypes (Kloosterman et al. 2011b, 2012; Chiang et al 2012). The nature of chromosome breakage and reassembly appears similar for cancer and germline, although germline chromothripsis rearrangements have substantially less breakpoints and display a more balanced state (Kloosterman et al. 2011b, 2012; Chiang et al 2012). Both differences are likely due to strong selective pressure against catastrophic DNA damage during embryonic development. Alternatively, they could reflect differences in chromothripsis mechanisms (Zhang et al. 2013; Kloosterman and Cuppen 2013).
5 The Mechanism Behind Chromothripsis
Although chromothripsis has been found in many different cancer types and appears a major mutational force driving tumor development, the precise origin of chromothripsis is only beginning to be understood. The vast majority of chromothripsis breakpoint junctions are characterized by microhomology of 2–4 nt (Stephens et al. 2011; Rausch et al. 2012; Morrison et al. 2014). This is consistent with repair of the DNA fragments by nonhomologous end-joining (NHEJ) (Moore and Haber 1996).
Several hypotheses and experimental data have shed light on possible causes of chromothripsis (Stephens et al. 2011; Zhang et al. 2013; Kloosterman et al. 2012; Holland and Cleveland 2012; Forment et al. 2012; Maher and Wilson 2012). The most compelling explanation concerns the formation of micronuclei that capture (part of) one or multiple chromosomes following chromosome segregation errors (Crasta et al. 2012; Hatch et al. 2013). This model for chromothripsis formation provides an elegant explanation for the clustering of rearrangements to one or a few chromosomes, because these are physically separated during DNA damage formation and repair, but can rejoin the other chromosomes in the nucleus during subsequent cell divisions (Crasta et al. 2012; Hatch et al. 2013). It has been shown that repair of DNA damage is defective and/or delayed in micronuclei due to defects in DNA damage response signaling (Crasta et al. 2012; Terradas et al. 2009). Chromosomes in micronuclei showed γ-H2AX foci, but lacked efficient recruitment of its downstream components of the DNA damage response, leading to extended persistence of γ-H2AX foci in the micronuclei (Crasta et al. 2012). Furthermore, DNA replication in micronuclei is asynchronous with the primary nucleus as many micronuclei show DNA replication in G2-phase (Crasta et al. 2012). Premature chromosome compaction (PCC) is a well-known mechanism that can lead to pulverization of chromosomes (Johnson and Rao 1970; Sperling and Rao 1974). If micronuclei enter mitosis before completion of DNA replication PCC can occur, leading to the occurrence of massive amounts of DNA double-stranded breaks (DSBs) (Crasta et al. 2012; Donley and Thayer 2013). This damage occurs in the first cell cycle following micronuclei formation (Crasta et al. 2012). Indeed, chromosome paintings of micronucleated cells demonstrated small fragments from one or two chromosomes (Crasta et al. 2012). A study by Hatch et al. (2013) uncovered a possible explanation for DNA damage to chromosomes contained in micronuclei. The authors showed a strong correlation between micronuclear envelope breakdown and the occurrence of massive DNA damage, although it remains unknown how nuclear envelope breakdown exactly leads to this damage (Hatch et al. 2013). Recent work has now demonstrated that chromosomes captured in micronuclei can undergo massive genomic rearrangements, including all the known hallmarks of chromothripsis as observed in cancer genomes (Zhang et al. 2015), thus providing experimental proof for a mechanism causing chromothripsis.
Although the micronucleus model has now been tested experimentally, other possible causes of chromothripsis may exist, including the influence of exogenous sources of DNA damage during mitosis. It has been suggested that free radicals and ionizing radiation can trigger chromothripsis rearrangements during mitosis when the chromosomes are highly condensed and DNA damage signaling is suppressed (Stephens et al. 2011; Zhang et al. 2011, 2013; Kloosterman and Cuppen 2013; Holland and Cleveland 2012; Maher and Wilson 2012). External damage occurring during this state of the cell cycle could possibly slice through multiple, closely located, segments of one or a few chromosomes, thereby explaining the localized character of chromothripsis (Stephens et al. 2011; Zhang et al. 2013).
Finally, it has been suggested that the occurrence of repeated BFB-cycles associated with telomere attrition could lead to chromothripsis rearrangements (Stephens et al. 2011; Sorzano et al. 2013). Telomere attrition leads to dicentric chromosomes, which form an anaphase bridge when the centromeres are pulled to opposite daughter cells during anaphase. The dicentric chromosome subsequently breaks at a random location between the two centromeres and fragments of this break are inherited by the daughter cells (Sorzano et al. 2013). It has been suggested that the dicentric chromosome may acquire the massive DNA damage seen in chromothripsis at the cleavage furrow during cytokinesis in a one-off event (Holland and Cleveland 2012) or that successive BFB-cycles, over the course of multiple cell divisions, are responsible for chromothripsis rearrangements (Sorzano et al. 2013).
Repeated BFB-cycles can explain the clustering of rearrangements near telomeres. However, BFB-cycles characteristically lead to multiple amplified copy number states, which is inconsistent with the two copy number states typically detected in chromothripsis. Furthermore BFB-cycles lead to an enrichment of inverted-type rearrangements, while rearrangement orientations in chromothripsis are randomly distributed (Bignell et al. 2007; Korbel and Campbell 2013; Zhang et al. 2013). A more plausible suggestion is the incorporation of lagging dicentric chromosomes into a micronucleus, rather than the nucleus of one of the daughter cells. In this way, telomere attrition may facilitate the occurrence of chromothripsis by inducing micronuclei formation (Holland and Cleveland 2012; Li et al. 2014). In acute lymphoblastic leukemia (ALL) the occurrence of dicentric chromosomes due to BFB-cycles or a germline Robertsonian translocation preceded chromothripsis, possibly through micronuclei formation after the formation of an anaphase bridge by the lagging chromosome (Li et al. 2014).
6 Other Types of Complex Genomic Rearrangements in Cancer Genomes
Prostate cancers exhibit patterns of complex rearrangements related to chromothripsis, characterized by chains of translocations and deletions, that occur in a highly interdependent manner (Fig. 4a). This phenomenon has been termed chromoplexy (from the Greek pleko, which means to weave or braid) (Baca et al. 2013). Chromoplexy chains typically contain three to over 40 rearrangements, involving one to up to ten chromosomes. A key characteristic is the occurrence of ‘deletion bridges’, small deletions between fusion junctions of translocation rearrangements in a subset of rearrangements, although chromoplexy in general seems to be a relatively copy-neutral process, when compared to chromothripsis (Zhang et al. 2013; Baca et al. 2013). Chromoplexy has been detected in 88 % of prostate tumors and frequently accounts for the dysregulation of prostate cancer genes, often disrupting multiple genes coordinately. Strikingly, two or more separate chromoplexy chains were detected in 63 % of prostate tumors (Baca et al. 2013). Where chromothripsis typically leads to a very high number of rearrangements in one event, multiple chromoplexy events can occur in successive cell cycles (Fig. 4b) (Baca et al. 2013). Chromoplexy rearrangements are usually not as highly localized as chromothripsis, as chromothripsis typically involves only one or a few chromosomes, whereas chromoplexy can involve up to ten chromosomes (Stephens et al. 2011; Baca et al. 2013). Furthermore, the total number of rearrangements in chromoplexy is smaller than for chromothripsis. Also, the relatively high number of (large) deletions seen in chromothripsis is not found in chromoplexy (Baca et al. 2013). Thus, chromoplexy could be regarded as a phenomenon involving rearrangements of intermediate complexity. In ETS + prostate cancer, well-known ERG fusions (e.g. TMPRSS2-ERG) frequently result from chromoplexy (58 %) (Baca et al. 2013). This clearly emphasized the oncogenic capability of chromoplexy in prostate tumors.
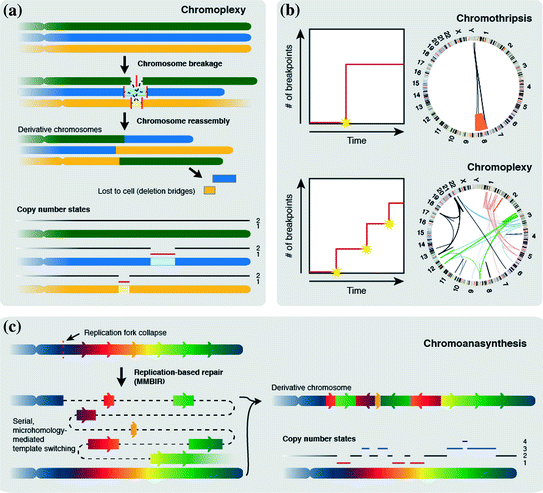
Fig. 4
Other mechanisms for complex rearrangements. a Chromoplexy. Multiple chromosomes undergo one or multiple DSBs (solid lines indicate breaks). Chained translocations are formed when chromosomes are reassembled with deletion bridges spanning translocations in some cases. In general, chromoplexy leads to fewer copy number alterations when compared to chromothripsis. b Differences between chromothripsis and chromoplexy. Chromothripsis generates a large number of breakpoints in a one-off catastrophic event. Chromoplexy leads to fewer breakpoints per event, but multiple chromoplexy chains can be formed in one cell in subsequent cell cycles. Due to this, chromothripsis usually leads to extreme clustering of breakpoints, whereas chromoplexy breakpoint clustering is less extreme. c Chromoanasynthesis is a replication-dependent process. Replication fork collapse leads to serial microhomology-mediated template switching leading to the formation of a derivative chromosome showing inversions and deletions, but also duplications and triplications of genomic regions
Like chromothripsis, each chromoplexy chain is suggested to form during a one-off catastrophic event, which is supported by the fact that both ends of each break are involved in chained translocations. A mechanism for the occurrence of this phenomenon remains to be established, but chromoplexy in ETS + tumors involves highly transcribed regions, which are colocalized in interphase nuclei, suggesting a mechanistic coupling of chromosome rearrangement and transcription (Baca et al. 2013).
An alternative category of complex genomic rearrangements was described in patients with developmental delay and cognitive anomalies (Liu et al. 2011). Chromoanasynthesis (anasynthesis for reconstruction) is characterized by interspersed changes in copy number, including duplications, triplications and deletions, combined with translocations and inversions (Fig. 4c). Like chromothripsis, chromoanasynthesis rearrangements are highly localized, usually involving only a single chromosome. Breakpoint junctions of chromoanasynthesis rearrangements frequently show microhomology or templated sequence insertions (54–1542 bp) (Liu et al. 2011). Despite the similarities between chromothripsis and chromoanasynthesis there are marked differences concerning rearrangement clustering and copy number changes (Zhang et al. 2013; Kloosterman and Cuppen 2013; Holland and Cleveland 2012). Chromoanasynthesis is characterized by multiple copy number states on a single chromosome due to a combination of deletions, duplications and triplications, as opposed to chromothripsis, where deletions are frequently observed, but typically no duplications or triplications are found (Zhang et al. 2013; Liu et al. 2011). Furthermore, detailed analysis of chromoanasynthesis breakpoint junctions revealed that these lack the typical double-strand break signatures that are found in chromothripsis (Kloosterman and Cuppen 2013).
The templated insertions and microhomology found at breakpoint junctions in chromoanasynthesis rearrangements are a signature of replication-based mechanisms, specifically microhomology-mediated breakage-induced repair (MMBIR) (Liu et al. 2011; Carvalho et al. 2009; Zhang et al. 2009). MMBIR is initiated by a broken DNA replication fork and leads to the incorporation of templated sequences of rearranged segments through serial, microhomology-mediated template switching, explaining the occurrence of multiple copy number states (Fig. 4c) (Liu et al. 2011; Hastings et al. 2009).
7 Methods for Identificaton of Complex Genomic Rearrangements in Cancer Genomes
Despite the major technical advances in the past decades (Box 1), the detection of genomic rearrangements remains complex due to both technical issues related to next-generation DNA sequencing, such as short sequence reads and small library insert size, and biological factors, such as the occurrence of (highly) repetitive regions, overlapping SVs and the complexity of breakpoints. Although different types and sizes of simple SVs can be detected relatively straightforward, the distinction of chromothripsis from other complex rearrangement types is challenging. Several bioinformatics tools have been developed to specifically detect chromothripsis or chromoplexy rearrangements (Baca et al. 2013; Cai et al. 2014; Govind et al. 2014; Malhotra et al. 2013; Kim et al. 2013). An essential part of these tools is their definition of criteria to distinguish each type of complex genomic rearrangement. These criteria are not entirely fixed across different studies, leading to differences in sensitivity for detection of complex rearrangement types (Table 1).
Table 1
Variation in criteria and methods used for chromothripsis and chromoplexy detection
Study (type) | Criteria | Technique (sample size) | Detection tool |
|---|---|---|---|
Stephens et al. (2011) (Chromothripsis) | (1) Tens to hundreds of genomic rearrangements that show geographic localization within the genome (2) Pronounced clustering of breakpoints on chromosomes or chromosome arms (3) Rearranged regions show frequent oscillations between two and occasionally three copy number states (4) Regions with copy number 1 are caused by a series of complex rearrangements that span the involved region and not by simple deletions (5) Heterozygosity is retained in regions with higher copy number (6) The complex rearrangements represent all four intrachromosomal breakpoint orientations in approximately equal numbers (7) The two conjoined fragments of chromosomes at each breakpoint fusion are often not located in the proximity of each other on the reference genome (8) Rearrangements involve a single parental copy of the chromosome (9) Double minute chromosome may form | Paired-end sequencing, SNP array (776) | In-house SV/CNA detection tools |
Magrangeas et al. (2011) (Chromothripsis) | Identical to criteria set by Stephens et al. (2011) | SNP array (764) | Partek Genomic Suite |
Rausch et al. (2012) (Chromothripsis) | At least 10 changes in segmental copy number involving two or three copy-number states per chromosome | Paired-end/long-mate-pair sequencing, SNP array (605) | PEMer tool and in-house tools |
Kim et al. (2013) (Chromothripsis) | (1) At least 10 CNAs localized to one chromosome (2) Occurrence of breakpoints is random e.g. the size of neighboring segment is similar | aCGH, SNP array (8227) | In-house statistic tool |
Malhotra et al. (2013) (Chromothripsis) | Breakpoints are assigned to the same breakpoint cluster if: (1) Breakpoints are located within 100 kb of each other in the reference genome (2) Different loci share breakpoint calls, indicating interconnection between breakpoints Breakpoint clusters are defined as complex and resulting from a one-off event when: (3) No more than three copy number states are detected (4) A maximum of one amplified copy number state exceeding four predicted copies is present in the region and this is not a focal amplification of a contiguous amplified region. Chromothripsis: (5) At least 10 or more clustered breakpoints and copy number profiles are present that meet all four criteria described above. | Paired-end sequencing, de novo assembly (64) | HYDRA-MULTI and in-house tools |
Zack et al. (2013) (Chromothripsis) | (1) Unexpectedly large number of CNAs on one chromosome compared to the CNA frequency of the sample (2) CNAs on a single chromosome are highly localized, discordant with what is expected by chance (3) CNAs lead to copy number changes of +1 or −1 and are non-overlapping | SNP array (4934) | In-house method, to be published |
Cai et al. (2014) (Chromothripsis) | (1) Significant clustering of breakpoints (2) Segmental copy number changes | Paired-end/mate-pair sequencing, whole-genome sequencing, SNP array, aCGH (18,394) | Scan-statistic based algorithm |
Govind et al. (2014) (Chromothripsis) | (1) Genome localization: low number of chromosomes involved in complex rearrangements (2) Chromosome localization: highly localized SV clustering on chromosome (3) Low number of copy number states, but high number of copy number oscillations (4) Translocation clustering: high number of localized translocations (5) Insertions at translocation breakpoints: high fraction of translocations where short insertions are found (6) High retained heterozygosity in areas between CNVs (7) Presence of a non-synonymous TP53 mutation (does currently not influence chromothripsis score) | Whole-genome sequencing (21) | Various tools for SV detection, ShatterProof for detection of chromothripsis |
Baca et al. (2013) (Chromoplexy) | ChainFinder determines whether genomic rearrangements where formed in an interdependent manner and assigns breakpoints that where formed interdependently to the same (rearrangement) chain (e.g. cycle) (1) Two breakpoints are assigned to the same chain if the p-value for the independent generation of the breakpoints is rejected with a false-discovery rate >10−2. (2) All scenarios are considered for one or more rearrangements in a cycle to occur independently. Only if each scenario is rejected with a family-wise error rate <10−2, all rearrangements in a cycle are assigned to the same chain | NGS, paired-end sequencing (57) | dRanger algorithm for detection of SVs, ChainFinder for detection of chromoplexy |
A new software tool, termed ShatterProof, was developed to standardize detection and quantification of chromothripsis (Govind et al. 2014). ShatterProof identifies highly mutated regions and subsequently scans these for chromothripsis based on seven variables (Table 1): (1) Genome localization: a high score indicates a much higher SV density on the investigated chromosome than on other chromosomes; (2) Chromosome localization: a high score indicates a high SV density in the region compared to the SV density of the total chromosome; (3) Aberrant CNVs: The presence of a low number of copy number states but a high number of copy number oscillations leads to a high score; (4) Translocation clustering: regions that show a high number of localized translocations are given a high score; (5) Insertions of translocation breakpoints: regions with a high fraction of translocations where short insertions are found receive a high score; (6) Amount of retained heterozygosity: regions that retained heterozygosity in areas between CNVs receive a high score; (7) TP53 mutation; the presence of non-synonymous TP53 mutations is reported back, but does not influence the chromothripsis score. An overall chromothripsis score is determined per region and reported back to the user. The higher the overall score, the more chromothriptic the region is. Although this tool provides a standardized pipeline for detection of chromothripsis, ShatterProof uses SV calls generated by other tools that have varying outcomes, thereby also influencing the outcome of ShatterProof, affecting the robustness of the tool.
Other tools for chromothripsis detection required either genome sequencing data (Malhotra et al. 2013), or copy number profiles from arrayCGH or SNParray (Cai et al. 2014; Kim et al. 2013). The differences between each of these tools mainly concern assumptions on the numbers of breaks involved, the numbers of copy number states allowed and the clustering of breakpoints within chromosomes (Table 1).
Baca et al. (2013) developed an algorithm specifically for detection of chromoplexy chains, termed ChainFinder. This algorithm identifies genomic rearrangements and their associated deletions that appear to have arisen interdependently from combined analysis of somatic breakpoints junctions and segmented copy number profiles. In order to do so, ChainFinder first identifies potential deletion bridges and subsequently performs a statistical analysis of all closely neighboring breakpoint pair distances to identify chain-like rearrangement patterns. Next, ChainFinder determines the probability of observing the two detected fusion breakpoints independently (i.e., not formed in the interdependent manner as breakpoints formed in a chain). Only breakpoints fulfilling the criteria for interdependent formation are assigned to chains (Table 1). It has been suggested that ChainFinder may be used in a broader sense to distinguish chromoplexy and chromothripsis events from other non-associated rearrangements on the same chromosome or its other parental counterpart (Zhang et al. 2013).
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


