Abbreviations
- ACTH Adrenocorticotropic hormone
- AKR1C2 3α-Hydroxysteroid dehydrogenase-3 (3α-HSD-3)
- AMH Anti-Müllerian hormone
- BPES Blepharophimosis-ptosis-epicanthus inversus syndrome
- CAH Congenital adrenal hyperplasia
- CIS Carcinoma in situ
- CBX2 Chromobox homolog 2, homolog of mouse m33
- CMPD1 Campomelic dysplasia
- DAX1 DSS-AHC-critical region on the X chromosome gene 1
- DHEA Dehydroepiandrosterone
- DHH Desert hedgehog
- DHT Dihydrotestosterone
- DMRT1 Double sex Mab3-related transcription factor gene 1
- DSS Dosage-sensitive sex reversal
- DSD Disorders of sexual development—congenital conditions in which development of chromosomal, gonadal, or anatomic sex is atypical
- 46,XX DSD Previously designated female pseudohermaphrodite
- 46,XY DSD Previously designated male pseudohermaphrodite
- FISH Fluorescent in situ hybridization
- FGF-9 Fibroblast growth factor-9
- FOXL2 Forkhead box L2
- FSH Follicle-stimulating hormone
- GATA-4 Member of the GATA family of zinc finger transcription factors
- GH Growth hormone
- GnRH Gonadotropin-releasing hormone
- hCG Human chorionic gonadotropin
- HMG High mobility group
- 17β-HSD-3 17β-Hydroxysteroid dehydrogenase-3
- ICSI Intracytoplasmic sperm injection
- IGF Insulin-like growth factor
- LH Luteinizing hormone
- MAMLD1 Mastermind-like domain containing 1
- MAP3K4 Mitogen-activated protein kinase kinase kinase 4
- Ovotesticular DSD Previously designated true hermaphrodism
- PAR Pseudoautosomal region
- POR P450 oxidoreductase
- PRKX Protein kinase X linked
- PRKY Protein kinase Y linked
- RFLP Testriction fragment length polymorphism
- RSPO1 R-spondin1
- SF-1 Steroidogenic factor-1
- SHOX Short stature homeobox gene on the X chromosome
- SOX3 SRY-like HMG box-3
- SOX9 SRY-like HMG box-9
- SRY Sex-determining region Y gene
- StAR Steroidogenic acute regulatory protein
- TSPY Testes-specific protein-Y encoded
- WAGR Wilms tumor-aniridia-genital anomalies-mental retardation syndrome
- WNT4 Human homolog of Drosophila wingless gene
- WT1 Wilms tumor gene
- XIST X-inactive specific transcript gene
Disorders of Sex Determination and Differentiation: Introduction
Advances in developmental and cell biology, molecular genetics, experimental embryology, steroid biochemistry, and methods of evaluation of the interaction between the hypothalamus, pituitary, and gonads as well as behavioral science have helped clarify problems of sexual determination and differentiation. Anomalies may occur at any stage of intrauterine development of the hypothalamus, pituitary, gonads, and genitalia and lead to gross ambisexual development or to subtle abnormalities that do not become manifest until sexual maturity is achieved.
Human Sex Differentiation
The normal human diploid cell contains 22 autosomal pairs of chromosomes and two sex chromosomes (two Xs, or one X and one Y). When arranged serially and numbered according to size and centromeric position, they are known as a karyotype. Advances in the techniques of staining chromosomes permit positive identification of each chromosome by its unique banding pattern. A technique called fluorescence in situ hybridization (FISH) is particularly useful in identifying quickly both sex chromosomes, mosaicism and structural abnormalities involving the sex chromosomes, the presence or absence of SRY (the testes determining gene on the Y chromosome), and other deleted genes (Figure 14–1). High-resolution chromosome banding and painting techniques provide precise identification of each chromosome.
Figure 14–1
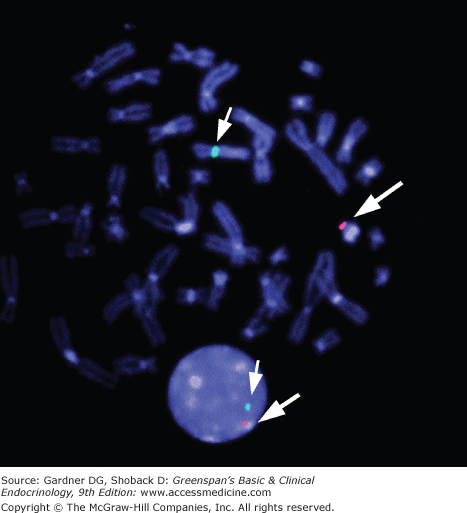
FISH (fluorescent in situ hybridization) for SRY in metaphase and interphase cells. Image illustrates localization of the SRY probe on the distal short arm of the Y chromosome (Yp11.3) in spectrum orange. The probe for the centromere region of the X chromosome is shown in spectrum green. Note the presence of both probes in the interphase cell below.
(Reproduced, with permission, from Grumbach MM, Hughes IA, Conte FA. Disorders of sex differentiation. In: Larsen PR, et al, eds. Williams Textbook of Endocrinology. 10th ed. WB Saunders; 2003.)
Studies in animals as well as humans with abnormalities of sexual differentiation indicate that the sex chromosomes (the X and Y chromosomes) and the autosomes harbor genes that influence sex determination and differentiation by causing the bipotential gonad to develop either as a testis or as an ovary. Two intact and normally functioning X chromosomes, in the absence of a Y chromosome or its SRY gene, lead to the formation of an ovary under normal circumstances, whereas a Y chromosome (whose SRY gene is intact) or the translocation of SRY, the testis-determining gene on the short arm of the Y chromosome, to an X chromosome or autosome leads to testicular organogenesis.
In humans, there is a marked discrepancy in size between the X and Y chromosomes. Gene dosage compensation is achieved in all persons with two or more X chromosomes in their genetic constitution by partial inactivation of all X chromosomes except one. This phenomenon is thought to be a random process (except when a structurally abnormal X chromosome is present) that occurs in each cell in the late blastocyst stage of embryonic development, during which either the maternally or the paternally derived X chromosome undergoes heterochromatinization. A result of this epigenetic process is formation of an X chromatin body (Barr body) in the interphase cells of persons having two or more X chromosomes. In patients with two or more X chromosomes, the maximum number of Barr bodies (partially inactivated X chromosomes) seen in interphase cells will be one less than the number of X chromosomes in the karyotype. A gene termed XIST (X-inactive specific transcripts) is located in the X inactivation center at Xq13.2 on the paracentromeric region of the long arm of the X chromosome. XIST is expressed only by the inactive X chromosome. The XIST gene encodes a large RNA that appears to coat the X chromosome and facilitates inactivation of selective genes on the X chromosome.
The distal portion of the short arm of the X chromosome escapes inactivation and has a short (2.5-megabase [mb]) segment homologous to a segment on the distal portion of the short arm of the Y chromosome (Figures 14–2 and 14–3). This segment is called the pseudoautosomal region (PAR); it is these two limited regions of the X and Y that pair during meiosis, undergo obligatory chiasm formation, and allow for exchange of DNA between these specific regions of the X and Y chromosomes. At least 10 genes have been localized to the pseudoautosomal region on the short arm of the X and Y chromosomes. Among these are a gene whose deletion results in the neurocognitive defects observed in Turner syndrome and a gene for short stature, SHOX (short stature homeobox gene), which is expressed in bone. A mutation or deletion of SHOX on either the X or Y chromosome is associated with “idiopathic” short stature as well as dyschondrosteosis (Leri-Weill syndrome). Homozygous mutations of this gene are associated with a more severe form of short stature, Langer mesomelic dwarfism. A pseudoautosomal region has also been described for the distal ends of the long arms of the X and Y chromosomes (see Figures 14–2 and 14–3). The pseudoautosomal region of the long arms of the X and Y chromosomes contains genes that are mostly growth factors and signaling molecules. The Y chromosome (see Figure 14–3) represents only 2% of the human genomic DNA and is about 60 mb in length. It is unique in that it contains few active genes compared to the X and autosomal chromosomes and has a large apparently noncoding heterochromatic region. It contains at least two genes affecting growth, the SHOX gene in the PAR and the growth control gene on the Y chromosome (GCY). The euchromatic region of the short arm of the Y chromosome contains the SRY gene (the male determining factor) distal to the PAR region at Yp11.3. The short arm of the Y chromosome contains genes that when deleted produce the phenotype stigma of Turner syndrome. The pericentromeric region of the Y is the locus for the TSPY (testes-specific protein Y encoded) genes which predispose to gonadoblastoma formation in the presence of dysgenetic testicular development. The PRKY gene is homologous to a gene PRKX (protein kinase X linked) on the X chromosome and is the locus for Y to X translocation observed in 46,XX SRY-positive males. The euchromatic region of the long arm of the Y chromosome has genes that when deleted result in azoospermia.
Figure 14–2
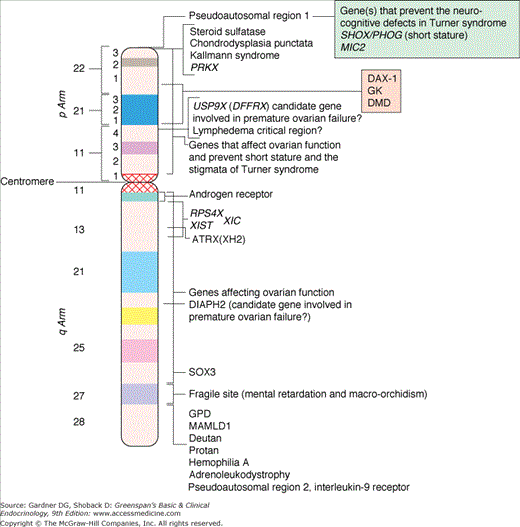
Diagrammatic representation of G-banded X chromosome. Selected X-linked genes are shown (ATRX, α-thalassemia, X-linked mental retardation; DAX1, DSS-AHC-critical region on the X chromosome gene 1; DIAPH2, human homolog of the Drosophila diaphanous gene; DMD, duchenne muscular dystrophy; GK, glycerol kinase; GPD, glucose-6-phosphate dehydrogenase; MAMLD1, mastermind-like domain-containing 1; MIC2, a cell surface antigen recognized by monoclonal antibody 12E7; PRKX, a member of the cAMP-dependent serine-threonine protein kinase gene family [illegitimate X-Y interchange occurs most frequently between PRKX and PRKY]; RPS4X, ribosomal protein S4; SHOX, short-stature homeobox gene; SOX3, SRY-like HMG box-3; USP9X, human x-linked homolog of the drosophila fat facets-related gene [DFFRX]; XIC, X-inactivation center; XIST, Xi-specific transcripts).
(Reproduced, with permission, from Grumbach MM, Hughes IA, Conte FA. Disorders of sex differentiation. In: Larsen PR, et al, eds. Williams Textbook of Endocrinology. 10th ed. WB Saunders; 2003.)
Figure 14–3
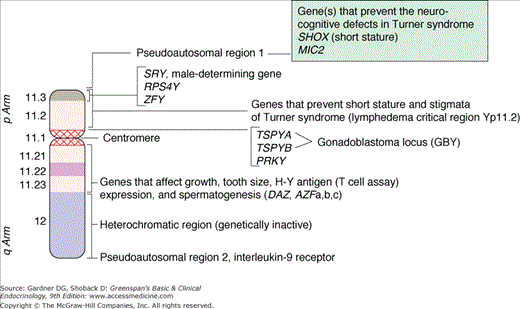
Diagrammatic representation of a G-banded Y chromosome (AZF, azoospermic factor; DAZ, deleted in azoospermia; MIC2, gene for a cell surface antigen recognized by monoclonal antibody 12E7; PRKY, a member of the cAMP-dependent serine-threonine protein kinase gene family; RPS4Y, ribosomal protein S4; SHOX, short stature homeobox gene; SRY, sex-determining region Y; TSPYA,B, members of the testes-specific factor gene family; ZFY, zinc finger Y).
(Reproduced with permission from Grumbach MM, Hughes IA, Conte FA. Disorders of sex differentiation. In: Larsen PR, et al, eds. Williams Textbook of Endocrinology. 10th ed. WB Saunders; 2003.)
Heterozygous mutations and deletions of the Wilms tumor gene (WT1) located on 11p13 result in urogenital malformations as well as Wilms tumor. Knockout of the WT1 gene in mice results in apoptosis of the metanephric blastema and as a consequence, absence of the kidneys and gonads. Thus, WT1, a transcriptional regulator, appears to act on metanephric blastema early in urogenital development (Figure 14–4A). Dominant-negative point mutations of WT1 in human beings results in the Denys-Drash and Frasier syndromes, whereas a contiguous deletion of the gene and surrounding DNA results in Wilms tumor, aniridia, ambiguous genitalia, and mental retardation—the WAGR syndrome.
Figure 14–4A

Major genes involved in sex determination from the intermediate mesoderm to the bipotential gonad. (DMRT1, doublesex and mab-3-related transcription factor 1; PGC, primordial germ cell [arise from an extragonadal site]; SF-1, steroidogenic factor 1; SOX9, SRY-like HMG-box 9; WT1, Wilms tumor suppressor gene-1) The bipotential gonad contains the precursor cells which differentiate into the supporting cells, steroid hormone-producing cells, and germ cells as shown.
Steroidogenic factor-1 (SF-1) is a nuclear receptor involved in transcriptional regulation of many genes, including those that are involved in gonadal development, adrenal development, steroid synthesis, and reproduction (see Figure 14–4A). SF-1 is located on 9q33 and is expressed in the urogenital ridge as well as in steroidogenic organs. SF-1 is required for the synthesis of testosterone in Leydig cells; in Sertoli cells it regulates the anti-Müllerian hormone (AMH) gene. Homozygous knockout of the gene encoding Sf-1 (Sf-1 is the mouse homologue of mammalian SF-1) in mice results in apoptosis of the cells of the genital ridge that give rise to the adrenals and gonads and thus absence of gonadal and adrenal gland morphogenesis in both males and females. This gene has a critical role in the formation of all steroid-secreting glands (ie, adrenals, testes, and ovaries). Initial studies in humans with SF-1 mutations identified individuals with adrenal insufficiency, a 46,XY karyotype, complete gonadal dysgenesis, and the presence of müllerian derivatives, similar to the mouse phenotype. Subsequently, the spectrum of affected patients has included a range of 46,XY disorders of sexual development without adrenal insufficiency including hypospadias, anorchia, micropenis, complete gonadal dysgenesis, infertility in otherwise normal males, and ovarian failure in 46,XX females.
Haploinsufficiency of DMRT1 (double sex Mab3-related transcription factor gene 1)—a gene related to double sex in drosophila and Mab3 in Caenorhabditis elegans is a candidate for the abnormality in testis organogenesis in patients with 9p deletions (see Figure 14–4A). 46,XY patients invariably have the stigmata of the 9p syndrome (mental retardation, trigonocephaly, upslanting palpebral fissures, etc), as well as female or ambiguous external genitalia and Müllerian structures associated with streak gonads or dysgenetic testes. Recent data suggest that ovarian function may either be compromised or normal in 46,XX females with the 9p syndrome.
Ambiguous genitalia has also been reported in 46,XY patients with deletions of 10q. However, as yet a specific mutant gene(s) causing this DSD has not been identified on the 10q region.
Figure 14–4B
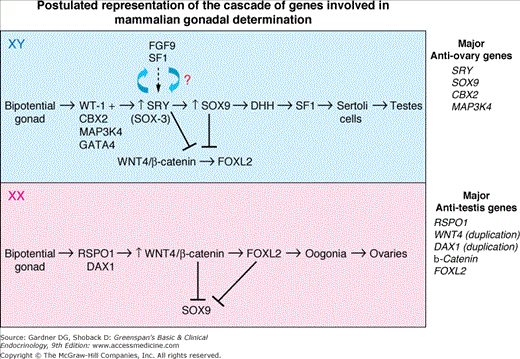
Opposing gene signals (differential activation) determines fate of primordial gonad as a testis or ovary. RSPO1, respondin 1; WNT4, human homolog of Drosophila wingless gene; FOXL-2, forkhead box L2; β-catenin, key-regulated effector of the WNT-signaling pathway. DAX-1 (DSS-AHC-critical region on the X chromosome gene 1) RSPO1 activates the WNT4/β-catenin canonical-signaling pathway which inhibits SOX9 expression and promotes ovarian differentiation. SRY upregulates SOX9 by binding to testicular enhancing elements in the SOX9 gene (TESCO), and both SRY and SOX9 (in mice) inhibit the β-catenin canonical-signaling pathway and promote Sertoli cell and consequent testicular development. A feed-forward loop involving SF-1 and FGF-9 has been demonstrated in mice which maintains SOX9 expression. FOXL2 “knockout” in mice causes gonadal sex reversal, i.e., ovary to testes suggesting that FOXL2 inhibits SOX9 expression. No human homozygous FOXL2 null mutations have been reported as yet. Duplication of DAX1 or WNT4 results in inhibition of testicular development and atypical genitalia—dysgenetic 46,XY DSD. MAP3K4, mitogen-activated protein kinase 4; CBX2, chromobox homologue 2. It has been suggested that CBX2 is upstream of SRY. More complete understanding of the exact function of MAP3K4 remains to be elucidated.
In studies of 46,XX males with very small Y-to-X translocations, a gene was localized to the region just proximal to the pseudoautosomal boundary of the Y chromosome (Yp11.3) (see Figure 14–3) which has been named SRY. Deletions or mutations of the human SRY gene occur in about 15% to 20% of 46,XY females with complete XY gonadal dysgenesis and 90% of 46,XX males have a Y to X translocation which includes the SRY gene. Compelling evidence that SRY is the testis-determining factor is the observation that transfection of the Sry gene into 46,XX mouse embryos results in transgenic 46,XX mice with testes and male sex differentiation.
The SRY gene encodes a DNA-binding protein that has an 80 amino acid domain similar to that found in high-mobility group (HMG) proteins. This domain binds to DNA in a sequence-specific manner (A/TAACAAT). It bends the DNA and thus is thought to facilitate interaction between DNA-bound proteins to affect the transcription of downstream genes. The human SRY gene has no intron; two nuclear localization sites, calmodulin and importin B are critical for the function of the gene. In mice, Sry facilitates the upregulation of Sox9 by binding to multiple enhancing elements in the Sox9 gene and along with Sf-1 establishes a feed-forward pathway resulting in upregulation of Sox9 and its continued expression in Sertoli cells. A feed–forward loop involving Sox9 and Fgf9 has also been demonstrated in the mouse but not yet in the human gonad. Both Sry and Sox9 in mice appear to inhibit the Rspo1 (R-spondin 1)-Wnt4-β-catenin- FOXL2 signaling pathway, facilitating Sertoli cell induction and testicular organogenesis and inhibiting ovarian development (see Figure 14–4B). In the absence of Sry, the Wnt4-β-catenin canonical pathway is stabilized and Sox9 is suppressed which results in ovarian development. Hence, it appears that Sry and Sox9 are repressors of a major ovarian determining pathway—Rspo1-Wnt4-β-catenin—which induces ovarian development along with the downstream gene—Fox L2 and other genes yet to be defined, as well as germ cells which are essential for ovarian development (but not testicular differentiation). Duplications of DAX1 or WNT4 in humans repress testicular development presumably by preventing the repression of β-catenin on its downstream gene FOXL2 by SRY and SOX9. Ovarian determination is a genetically controlled pathway and not, as previously thought, a default pathway.
Most of the mutations thus far described in 46,XY females with gonadal dysgenesis have occurred in the nucleotides of the SRY gene encoding the DNA-binding region (the HMG box) of the SRY protein. Mutations that affect DNA bending as well as nuclear transportation of the SRY protein have also been implicated in defective testicular development.
SOX9 has an HMG box that is more than 60% homologous to that of SRY. It is localized to 17q24.3-q25.1 and is expressed in the developing sex cords and, thereafter, in the Sertoli cells (see Figure 14–4B). It is also expressed in cartilage. Mutations in one allele of the SOX9 gene can result in a bone abnormality called campomelic dysplasia (CMPD) and XY gonadal dysgenesis with ambiguous genitalia in affected 46,XY males. Duplication of the SOX9 gene both in humans and mice results in sex reversal in XX individuals who are SRY negative. It appears that SOX9 is the critical gene acting downstream of SRY for Sertoli cell differentiation and consequent testicular development.
Desert hedgehog (DHH), a gene that codes for a signaling molecule located on chromosome 12q13.1 in humans, is an important gene in mammalian testes organogenesis and function. Mutations in DHH have been reported in individuals with 46,XY gonadal dysgenesis.
GATA4 GATA4 is a transcription factor which interacts with other proteins including SF-1 and FOG2 to regulate the expression of SRY. Recently, a heterozygous missense mutation has been described affecting three male siblings and resulting in 46,XY DSD and congenital heart disease.
A mutation or deletion of DAX1, which encodes a transcription factor, results in X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism in 46,XY males (see Figures 14–2, 14–4B). Deletion or mutation of the DAX1 gene in 46,XY individuals has not resulted in an abnormality of testicular differentiation in humans. However, XY gonadal dysgenesis has been reported in individuals with duplications of Xp21, a locus that contains the DAX1 gene on the X chromosome. Duplication of the DAX1 gene appears not to affect ovarian morphogenesis and function in 46,XX females. Thus, DAX1 dosage plays a critical role in testicular development and function (see Figure 14–4B).
WNT4 is a signaling molecule encoded by a gene located on the short arm of chromosome region 1 (1p35). Duplication of WNT4 has been associated with dysgenetic testicular development and ambiguous genitalia in 46,XY, SRY, positive individuals. Testis development is inhibited likely as a result of upregulation of DAX1 and stabilization of β-catenin-FOXL2 which would inhibit SOX9 expression in the developing gonad. 46,XY patients who have duplications of WNT4 exhibit a heterogenous phenotype varying from cryptorchidism alone to female external genitalia. WNT4 is expressed in the primordial ovary and is an essential signal for ovarian determination (see Figure 14–4B). In mice, Wnt4 expression in the ovary prevents the migration of steroid (androgen)-producing cells into the ovary, restrains the development of a testis-specific blood vessel (the coelomic vessel), and is critical for the development of the Müllerian ducts and the maintenance of germ cells.
A 46,XX female with absent Müllerian structures (atypical Mayer-Rokitansky-Küster-Hauser syndrome) has been reported who had a loss of function mutation in the WNT4 gene. Her phenotype was similar to that described in Wnt4-mutated mice. She had unilateral renal agenesis as well as clinical signs of androgen excess as manifested by severe acne requiring antiandrogen therapy. Wolffian duct development was not ascertained. Two other similar patients have been reported.
RSPO1 (R-spondin 1), a gene located in 1p34.3, appears to stabilize the expression of WNT4-β-catenin-FOXL2 and thus promotes ovarian determination (see Figure 14–4B). Mutations in RSPO1 cause 46,XX females to develop testes and male sexual development, as well as palmoplantar hyperkeratosis and a predisposition to squamous cell carcinoma of the skin. The fact that a mutation in RSPO1 can redirect gonadal development in a 46,XX individual from an ovary to a testes suggests that it is a critical determinant in ovarian determination in humans.
FOXL2 is a putative forkhead transcription factor, which does not cause a DSD in human. Heterozygous FOXL2 mutations in affected 46,XX human females causes premature ovarian failure with or without blepharophimosis. However, induced FoxL2 deletions in the ovary of the mouse lead to transdifferentiation of the ovary into testes.
A 46,XY phenotypic female with normal ovaries and a mutation in the CBX2 (chromobox homolog 2) gene, the human homolog of mouse m33, an ortholog of the Drosophila Polycomb gene, has recently been reported. It was postulated that this gene is upstream of SRY and that mutations in this gene prevent the repression of the ovarian gene pathway presumably by decreasing SRY/SOX9 expression enabling the expression of RSPO1-WNT4-β-catenin-FOXL2 and other ovarian determining genes.
Our lack of complete understanding of ovarian and testicular determination is illustrated in the study by Dumic et al who reported a familial cohort of 46,XY females. A 46,XY female was reported to have undergone spontaneous menarche and given birth to a 46,XY daughter with gonadal dysgenesis. Studies in this cohort have uncovered a mutation in a gene not previously known to be involved in the sex determination cascade—mitogen-activated protein kinase kinase kinase 4 (MAP3K4). Mutations in this gene involved in the mitogen-activated protein kinase signaling pathway cause a striking decrease in the expression of Sry and Sox9 in the developing mouse gonad resulting in sex reversal. Recently, mutations in MAP3K1 have been reported to 46,XY DSD leading to either partial or complete gonadal dysgenesis. Further studies in humans will be necessary to elucidate the exact mechanism which allows for repression of testicular development and the genesis of an apparently normal functioning ovary in SRY positive 46,XY individuals.
Testicular and Ovarian Differentiation
Figure 14–5
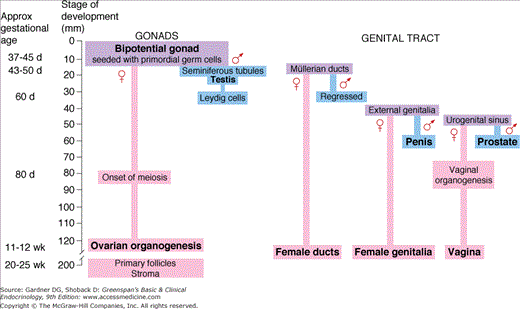
Schematic sequence of sexual differentiation in the human fetus. Note that testicular differentiation precedes ovarian differentiation.
(Reproduced, with permission, from Grumbach MM, Hughes IA, Conte FA. Disorders of sex differentiation. In: Larsen PR, et al, eds. Williams Textbook of Endocrinology. 10th ed. WB Saunders; 2003. Modified from Jost A. Hormone factors in sex differentiation of the mammalian fetus. Philo Trans R Soc Lond Biol. 1970;259:119.)
The mammalian gonad forms on the surface of the mesonephros within the genital ridge as a result of proliferation of the coelomic epithelium. Until the 12-mm stage (∼42 days of gestation), the embryonic gonads of males and females are indistinguishable. By 42 days, 300 to 1300 primordial germ cells have seeded the undifferentiated gonad from their extragonadal origin in the yolk sac dorsal endoderm. These large cells are the progenitors of oogonia and spermatogonia; lack of these cells is incompatible with further ovarian differentiation but not testicular differentiation. The undifferentiated gonad also contains supporting cells and steroidogenic cells. Under the influence of SRY/SOX9 and other genes that encode male sex determination, the gonad begins to differentiate as a testis at 43 to 50 days of gestation. The supporting cells differentiate into pre-Sertoli cells which organize into seminiferous tubules and surround the germ cells which undergo mitotic arrest. Leydig cells derived from steroidogenic cells are apparent by about 60 days, and differentiation of male external genitalia occurs by 65 to 77 days of gestation.
In the undifferentiated gonad destined to be an ovary, lack of further differentiation persists. At 77 to 84 days—long after differentiation of the testis in the male fetus—a significant number of germ cells enter meiotic prophase to characterize the transition of oogonia into oocytes, which marks the onset of ovarian differentiation from the undifferentiated gonads. The supporting cells become granulosa cells and steroidogenic cells become theca cells. Germ cell meiosis in the developing ovary is induced by retinoic acid signaling. Sry induces an enzyme which metabolizes retinoic acid to an inactive product and prevents meiosis of germ cells in the developing testis. Primordial follicles (small oocytes surrounded by a single layer of flat granulosa cells and a basement membrane) are evident after 90 days. Preantral follicles are seen after 6 months, and a population of fully developed oocytes with fluid-filled cavities and multiple layers of granulosa cells are present at birth. As opposed to the testes, there is little evidence of hormone production by the fetal ovary.
Figure 14–6
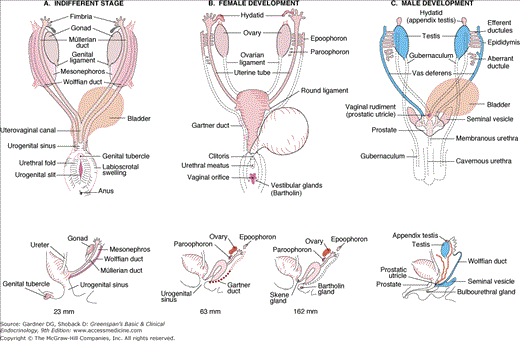
Embryonic differentiation of male and female genital ducts from Wolffian and Müllerian primordia. A. Indifferent stage showing large mesonephric body. B. Female ducts. Remnants of the mesonephros and Wolffian ducts are now termed the epoophoron, paroophoron, and Gartner duct. C. Male ducts before descent into the scrotum. The only Müllerian remnant is the testicular appendix. The prostatic utricle (vagina masculina) is derived from the urogenital sinus.
(Modified from Corning HK, Lehrbuch der Entwicklungsgeschichte des Menschen, JF Bergmann, Munich, 1921; and Wilkins L. The diagnosis and treatment of endocrine disorders. In: Charles C, ed. Childhood and Adolescence. 3rd ed. Thomas Publishers; 1965.)
By the seventh week of intrauterine life, the fetus is equipped with the primordia of both male and female genital ducts. The Müllerian ducts, if allowed to persist, form the uterine (fallopian) tubes, the corpus and cervix of the uterus, and the upper third of the vagina. The Wolffian ducts, on the other hand, have the potential for differentiating into the epididymis, vas deferens, seminal vesicles, and ejaculatory ducts of the male. In the presence of a functional testis, the Müllerian ducts undergo apoptosis under the influence of AMH, a dimeric glycoprotein secreted by fetal Sertoli cells. AMH acts locally to cause Müllerian duct repression ipsilaterally. Müllerian duct regression is the earliest indicator of male sex differentiation.
The gene for AMH encodes a 560 amino acid protein, whose carboxyl terminal domain shows marked homology with transforming growth factor-β (TGF-β) and the B chain of inhibin and activin. The gene is localized to the short arm of chromosome 19. AMH is secreted by human fetal and postnatal Sertoli cells and is used as a marker for the presence of these cells. SF-1 (an orphan nuclear receptor) in combination with WT1, SOX9, and GATA4 (Figure 14–7), regulates AMH gene expression. The human AMH receptor gene maps to the q13 band of chromosome 12. The receptor is similar to other type II receptors of the TGF-β family.
Figure 14–7

Hypothetical diagrammatic cascade of major genes involved in male sex differentiation
(DAX1, DSS-AHC-critical on theX chromosome gene 1; GATA4, transcription factor; SF-1, steroidogenic factor 1; SOX9, SRY homeobox gene 9; WT1, Wilms tumor suppressor gene). (Modified from Grumbach MM, Hughes IA, Conte FA. Disorders of sex differentiation. In: Larsen PR, et al, eds. Williams Textbook of Endocrinology. 10th ed. WB Saunders; 2003: 842-1002.)
The differentiation of the Wolffian duct is mediated by testosterone (T) secretion from the testis. SF-1 regulates steroidogenesis by the Leydig cell in the testes by binding to the promoter of the genes encoding P450 side chain cleavage enzyme (P450scc) and 17α-hydroxylase (P450c17). In the presence of an ovary or in the absence of a functional fetal testis, Müllerian duct differentiation occurs, and the Wolffian ducts involute (see Figure 14–6).
Figure 14–8
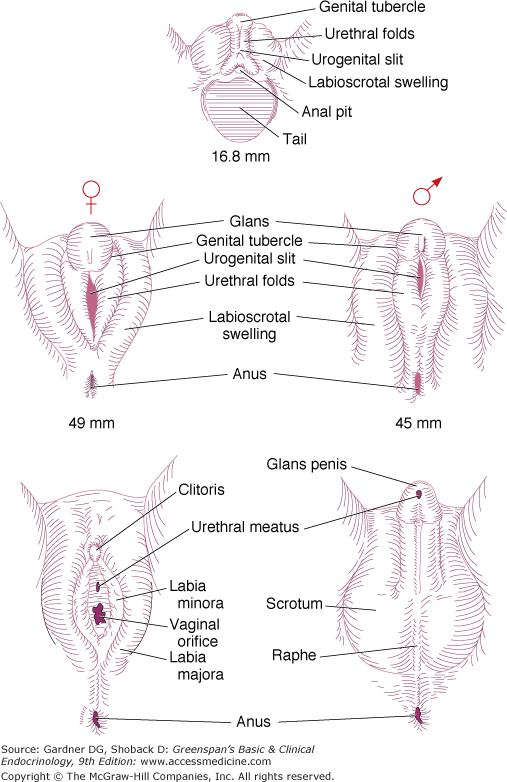
Differentiation of male and female external genitalia from bipotential primordia.
(Adapted from Spaulding MH. The development of the external genitalia in the human embryo. Contrib Embryol. [Carnegie Institute of Washington] 1921;13:69-88,. Reproduced with permission from Grumbach MM, Hughes IA, Conte FA. Disorders of sex differentiation. In: Larsen PR, et al, eds. Williams Textbook of Endocrinology. 10th ed. WB Saunders, 2003.)
Up to the eighth week of fetal life, the external genitalia of both sexes are identical and have the capacity to differentiate into the genitalia of either sex. Female sex differentiation occurs in the presence of an ovary or streak gonads or if no gonad is present (see Figure 14–8). The fetal ovary does not secrete sex steroids during gestation or AMH during the critical period of female differentiation of the genital ducts and external genitalia. Studies in the past have indicated that the fetal adrenal gland is 3β-HSD-2 (3 beta-hydroxysteroid dehydrogenase-2) deficient and secretes primarily DHEA-S (dehydroepiandrosterone sulfate) until late in gestation. DHEA-S is converted by the placenta to DHEA which is metabolized to T. T is aromatized to estrogen, thereby preventing the exposure of the female external genitalia to high levels of T/dihydrotestosterone (DHT). Placental aromatase activity is low from 8 to 12 weeks of gestation but increases markedly after the first trimester (Figure 14–9). Recent studies indicate that the normal female genitalia are protected from androgen exposure early in gestation (8-12 weeks), a stage when placental aromatase activity is relatively low. Sex differentiation of the external genitalia is occurring by the transient expression of 3β-HSD-2 in the fetal adrenal, resulting in cortisol secretion, and consequently relative suppression of both adrenocorticotropin (ACTH) and DHEA-S secretion with a resultant decrease in placental testosterone synthesis and secretion. Adrenally derived androgen levels appear to be the same during this period in both males and females. After 12 weeks of gestation, 3β-HSD-2 activity diminishes in the fetal adrenal which leads to an increase in ACTH and DHEA-S secretion. Testosterone is now converted by the increased placental aromatase to estrogen. In addition, androgen receptor activity diminishes in the labioscrotal folds, and excess androgen exposure after 12 to 14 weeks gestation results in only clitoromegaly, but not labioscrotal fusion. Thus, the transient expression of 3β-HSD-2 and the consequent ability of the fetal adrenal gland to transiently synthesize cortisol, as well as the decrease in androgen receptor activity in genital skin protects the female external genitalia from masculinization during the window of dimorphic differentiation of the external genitalia.
Figure 14–9
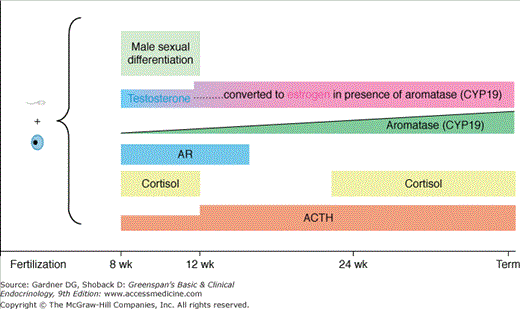
Sex differentiation of the external genitalia of males is induced by dihydrotestosterone (DHT) which is synthesized in the cells of the external genitalia from fetal testicular testosterone by the enzyme 5alpha-reductase-2. DHT may also be produced via the “back door pathway”. Male differentiation of the external genitalia occurs from 8 to 12 weeks of gestation when aromatase activity of the liver and placenta is relatively low. During this critical window of dimorphic sexual differentiation, the fetal adrenal transiently expresses 3β-HSD-2, resulting in adrenal secretion of cortisol and a consequent downregulation of fetal ACTH and T precursor (DHEA-S) secretion from the fetal adrenal as well as putative adrenal testosterone secretion. Thus, the female external genitalia are protected from T/DHT exposure and virilization during this period when aromatase activity is low. After 12 weeks of gestation, 3β-HSD-2 activity in the adrenal is extinguished until 24 weeks of gestation resulting in an increase in DHEA-S (T precursor) from the fetal adrenal. However, increased placental aromatase activity and a decrease in androgen receptor activity in the labioscrotal folds of 46,XX females occurs so that excess T/DHT exposure after 12 weeks of gestation results only in clitoromegaly.
(Modified from Hanley NA, Arlt W. The human fetal adrenal cortex and the window of sexual differentiation. Trends Endocrinol Metab. 2006;17:391.)
Differentiation of the external genitalia along male lines depends on the action of DHT, the 5α-reduced metabolite of T. In the male fetus, T is secreted by the Leydig cells, autonomously at first and later under the influence of human chorionic gonadotropin (hCG), and later fetal pituitary luteinizing hormone (LH). Masculinization of the external genitalia and urogenital sinus of the fetus results from the action of DHT, which is synthesized from T in the target cells by the enzyme 5α-reductase-2 as well as possibly secreted by the testes via the backdoor pathway (Figure 14–10). This pathway was first found in marsipials and then implicated in patients with P450 oxidoreductase (POR) deficiency. This backdoor pathway synthesizes DHT, a nonaromatized androgen, without utilizing T as a precursor. This pathway may play a signifcant role in male differentiation; however, further studies are necessary to define this.
Figure 14–10
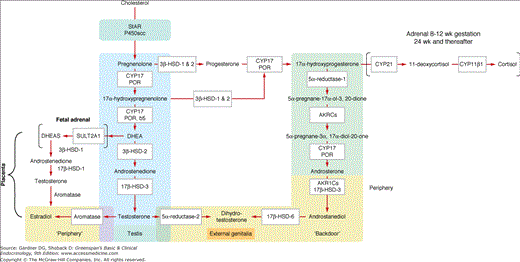
The enzymatic pathway of testosterone synthesis in the adrenal and testes is shown in blue on the left. A backdoor pathway first described by Wilson and Auchus from studies in marsupials is shown on the right. This pathway utilizes 17-OH progesterone to synthesize dihydrotestosterone independent of testosterone synthesis. The backdoor pathway as well as traditional testosterone synthesis by the fetal testis may be essential for normal male external genitalia differentiation. It has been suggested that virilization in 46,XX individuals with CAH (such as 21-hydroxylase, 11-hydroxylase, P450 oxidoreductase, and possibly 3β-HSD-2 deficiency) who have elevated levels of 17-OH progesterone in utero may involve DHT synthesis via the backdoor pathway. The fetal adrenal is relatively 3β-HSD-2 deficient from 12 to 24 weeks of gestation and DHEA-S is primarily secreted and converted in the liver to 16-OH DHEAS. DHEA-S is converted by placental sulfatase to DHEA which then is metabolized to T and subsequently to estradiol by placental aromatase. Similarly, 16OH-DHEAS is aromatized to estriol. Androstenedione can be directly converted to estrone by (3a-HSD-3 (AKRCs), 3-alpha-hydroxysteroid dehydrogenase-3; b5, cytochrome b5; 3β-HSD-1, 3-beta-hydroxysteroid dehydrogenase type 1; 3β-HSD-2, 3-beta-hydroxysteroid dehydrogenase type 2; 17β-HSD-1, 17-beta hydroxysteroid dehydrogenase type 1; 17β-HSD-3, 17-beta-hydroxysteroid dehydrogenase type 3; CY17, CYP21, 21 hydroxylase; CYP11B1, 11 hydroxylase;17hydroxylase; DHEA, dehydroepiandrosterone; P450scc, P450 side chain cleavage; POR, P450 oxidoreductase; StAR, steroidogenic acute regulatory protein; Sulf2A1, sulfatase).
DHT (as well as T) is bound to a specific protein receptor in the nucleus of the target cell (Figure 14–11). The transformed steroid-receptor complex dimerizes and binds with high affinity to specific DNA domains, initiating DNA-directed, RNA-mediated transcription. This results in androgen-induced proteins that lead to differentiation and growth of the cell. The gene that encodes the intracellular androgen-binding protein has been localized to the paracentromeric portion of the long arm of the X chromosome (see Figure 14–2). Of note, an X-linked gene controls the androgen response of all somatic cell types by specifying the androgen receptor protein. As noted previously, there is a decrease in androgen receptor activity in the labioscrotal folds after 12 to 14 weeks so that even with intense T/DHT stimulation, labioscrotal fusion does not occur although growth of the genital tubercle can continue resulting in clitoromegaly in females and penile enlargement in males.
Figure 14–11
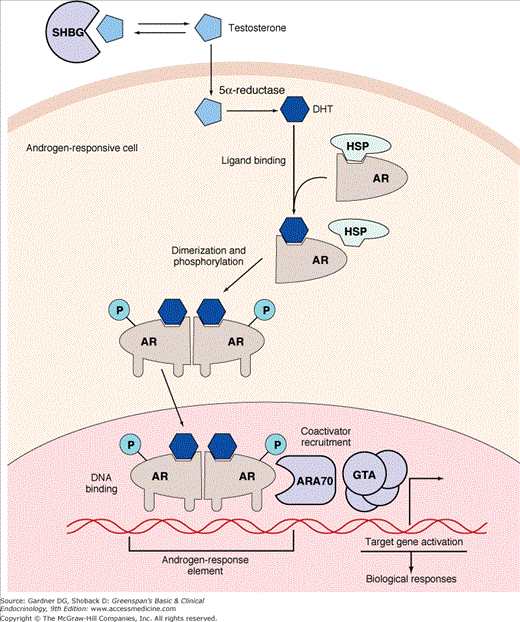
Diagrammatic representation of the mechanism of action of testosterone on target cells. Testosterone (T) circulates in association with sex hormone–binding globulin (SHBG) but dissociates to enter the cells, where it is either 5α-reduced to dihydrotestosterone (DHT) or aromatized to estradiol (E2). Dihydrotestosterone binds to the androgen receptor (AR) in the cytoplasm and activates it with the release of heat shock proteins (HSP). The activated AR complex is then translocated to the nucleus, where it binds as a dimer to specific hormone response elements of the DNA and, along with coactivators (eg, ARA), initiates transcription, leading to protein synthesis with consequent androgenic effects. (Redrawn and modified from Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34.)
Incomplete masculinization of the male fetus results from (1) impairment in the synthesis or secretion of fetal T and/or DHT (3α-HSD-3 deficiency) or the conversion of T to DHT (5α-reductase-2 deficiency, (2) deficient or defective androgen receptor activity, or (3) defective production and local action of AMH.
Psychosexual Differentiation
Psychosexual differentiation can be classified into four broad categories: (1) gender identity, defined as the identification of self as either male or female; (2) gender role (ie, those aspects of behavior in which males and females differ from one another in one’s culture at this time); (3) gender orientation, the choice of sexual partner; and (4) cognitive differences.
For much of the past 50 years, the prevailing dogma has been that newborns are born psychosexually neutral and that gender identity is imprinted postnatally by words, attitudes, family dynamics, and comparisons of one’s body with that of others. Gender identity had been hypothesized to be neutral at birth and not established until 12 to 18 months of age. During the last decade, this hypothesis has been rigorously challenged. It has become evident that prenatal exposure to T/DHT and possibly genes on the Y chromosome can influence gender identity in the individual with a DSD. A growing body of evidence indicates that gender identity is more plastic than previously thought. If, at puberty, discordant secondary sexual characteristics are allowed to mature, some individuals, especially those with 5α-reductase deficiency, 45,X/46,XY mosaicism, or 17β-HSD-3 deficiency, have had doubts about their gender identity and have chosen to change their assigned sex from female to male. However, in general, it has been demonstrated that most patients with DSD (except for those mentioned above) will accept their gender assignment even when it is not concordant with their sex chromosome constitution—a phenomenon which underscores both the complexity and plasticity of the processes that determine gender identity. This suggests that androgens have a facultative and not a deterministic role in gender identity. Androgens have a significant effect on childhood play behavior, maternal interest, and sexual orientation. It is apparent that both genes and hormones (nature) and environment and experience (nurture) are critical factors in the development and maintenance of gender identity. Further studies on patients with ambiguous genitalia, including long-term outcomes of the factors influencing gender identity, gender role, and sexual activity in DSD patients, are critical to furthering our understanding of the multifaceted complexities of the differentiation of gender identity and to support more evidence-based guidelines for decisions about sex assignment.
Classification and Nomenclature of Disorders of Sex Determination (and Differentiation)
| I. Sex chromosome DSD (disorders of gonadal differentiation) |
| A. Seminiferous tubule dysgenesis and its variants (Klinefelter syndrome): 46,XX testicular DSD |
| 1. 46,XX males, SRY positive |
| 2. 46,XX males, SRY negative |
| 3. SOX9 duplication |
| 4. RSPO1 mutation |
| 5. SOX3 mutation |
| B. Syndrome of gonadal dysgenesis and its variants (Turner syndrome) |
| C. Complete and incomplete forms of XX and XY gonadal dysgenesis |
| D. Individuals with both testicular and ovarian tissue: ovotesticular DSD (true hermaphroditisma) |
| E. 46,XY ovarian DSD–Fertile 46,XY female; MAP3K4 mutation |
| II. 46,XX DSD (female pseudohermaphrodisma) |
| A. Androgen induced |
| 1. Fetal source |
| a. Congenital virilizing adrenal hyperplasia (defective 21-hydroxylation, 11β-hydroxylation, 3β-hydroxysteroid dehydrogenase-2, P450 oxidoreductase (POR) |
| b. Glucocorticoid receptor mutation |
| 2. Fetoplacental source |
| a. P450 aromatase deficiency |
| b. P450 oxidoreductase deficiency (affecting aromatase) |
| 3. Maternal source |
| a. Iatrogenic |
| i. Testosterone and related steroids |
| ii. Certain synthetic oral progestagens |
| b. Virilizing ovarian or adrenal tumor |
| c. Virilizing luteoma of pregnancy |
| d. Congenital virilizing adrenal hyperplasia in motherb |
| B. Non–androgen-induced |
| 1. Disturbances in differentiation of urogenital structures associated with malformations of intestine and lower urinary tract (non–androgen-induced XX, DSD) (eg, cloacal anomalies, Müllerian agenesis [MURCS], vaginal atreasia, labial adhesions) |
| III. 46, XY DSD (male pseudohermaphrodisma) |
| A. hCG/LH receptor mutation (Leydig cell agenesis or hypoplasia) |
| B. Inborn errors of testosterone biosynthesis |
| 1. Enzyme deficits affecting synthesis of both corticosteroids and testosterone (variants of congenital adrenal hyperplasia) |
| a. StAR deficiency (congenital lipoid adrenal hyperplasia): side chain (P450scc) cleavage deficiency |
| b. 3β-Hydroxysteroid dehydrogenase-2 deficiency |
| c. P450c17 (17α-hydroxylase) deficiency |
| d. P450 oxidoreductase deficiency |
| e. 7-dehydrocholesterol reductase deficiency (Smith-Lemli-Opitz syndrome) |
| 2. Enzyme defects primarily affecting testosterone biosynthesis by the testes |
| a. P450c17 (17,20-lyase) deficiency |
| b. 17β-Hydroxysteroid dehydrogenase-3 deficiency |
| 3. Enzyme defects affecting DHT synthesis |
| a. 5α-Reductase-2 deficiency (pseudovaginal perineoscrotal hypospadias) (peripheral) |
| C. Defects in androgen-dependent target tissues |
| 1. End-organ resistance to androgenic hormones (androgen receptor and postreceptor defects) |
| a. Syndrome of complete androgen resistance and its variants (testicular feminization and its variant forms) |
| b. Syndrome of partial androgen resistance and its variants (Reifenstein syndrome) |
| c. Androgen resistance in infertile men |
| d. Androgen resistance in fertile men |
| D. Dysgenetic 46,XY DSD |
| 1. XY gonadal dysgenesis (incomplete) |
| 2. XO/XY mosaicism, SRY mutation structurally abnormal Y chromosome, Xp+ (DAX-1 dupl.), 9p−, 10q− |
| 3. Denys-Drash, Frasier syndrome (WT-1 mutation) |
| 4. WAGR syndrome (WT-1 deletion) |
| 5. Campomelic dysplasia (SOX9 mutation) |
| 6. SF-1 mutation |
| 7. WNT-4 duplication |
| 8. DHH (mutation) |
| 9. ATRX syndrome (XH2 mutation) |
| 10. ARX mutation |
| 11. CBX2 mutationc |
| 12. MAP3K4 mutation |
| 13. Testicular regression syndrome |
| 14. GATA4 mutation |
| 15. FOG2 mutation |
| E. Defects in synthesis, secretion, or response to AMH |
| 1. Female genital ducts in otherwise normal men—herniae uteri inguinale; persistent Müllerian duct syndrome |
| F. Environmental chemicals (endocrine disrupters) |
| IV. Unclassified forms of disorders of sexual development |
| A. In males |
| 1. Hypospadias: MAMLD-1 mutation |
| 2. Aphallia |
| 3. Ambiguous external genitalia in 46,XY males with multiple congenital anomalies |
| 4. Intrauterine growth retardation with incomplete masculinization of external genitalia |
| 5. Cloacal exstrophy |
| 6. Panoply of syndromes associated with incomplete masculinization of external genitalia (Robinow, Aarskog, hand-foot-genital, etc) |
| B. In females |
| 1. Absence or anomalous development of the vagina, uterus, and Müllerian ducts (Mayer-Rokitansky-Küster-Hauser [MRKH] syndrome) (WNT-4 mutation rare) |
An International Conference on Management of Intersex was held in Chicago, Illinois, in 2005 because of ethical issues and patient concerns about the venerable but antiquated terms intersex, pseudohermaphrodite, true hermaphrodite, and so forth, which were perceived by patients as pejorative. A new nomenclature was proposed and accepted. The term, disorders of sexual development (DSD), connoting all conditions in which chromosomal, gonadal, or anatomic sex are atypical, was coined. (See Lee PA et al for the complete consensus statement.)
DSDs are the result of abnormalities in complex processes that originate in genetic information on the X and Y chromosomes as well as on the autosomes. A 46,XY DSD (previously male hermaphrodite) is one whose gonads are exclusively testes in origin but whose genital ducts or external genitalia (or both) exhibit incomplete masculinization. A 46,XX DSD (previously female hermaphrodite) is a person whose gonadal tissue is exclusively ovarian in origin but whose genital development exhibits ambiguous or even a male appearance. Persons who possess both ovarian and testicular tissue were previously referred to as true hermaphrodites and now called ovotesticular DSD.
Klinefelter Syndrome and Its Variants: Seminiferous Tubule Dysgenesis—Sex Chromosome DSD
Klinefelter syndrome is one of the most common forms of primary hypogonadism and infertility in males. Surveys of the prevalence of 47,XXY fetuses by karyotype analysis of unselected newborn infants indicate a prevalence of about 1:700 newborn males. However, there is a striking disparity between the prevalence diagnosed at birth by karyotype screening and the frequency of Klinefelter syndrome identified in adults. It has been estimated that only 25% of individuals with Klinefelter syndrome are diagnosed during life and less than 10% in childhood. This most likely is due to the broad spectrum of phenotypes, testicular function, and psychosocial status found in patients with this syndrome. Ninety percent of affected patients have a 47,XXY sex chromosome constitution and an X chromatin-positive buccal smear, the rest demonstrate a variety of sex chromosome constitutions, including mosaicism. Virtually all of these variants have at least two X chromosomes and a Y chromosome in common, except for the rare group that has only an XX sex chromosome complement (3%-4%).
Hormonal levels are variable, and T levels may be in the low-normal to normal range. In addition, variability in phenotype can result in part from sex chromosome mosaicism with a normal XY cell line as well as variability in androgen sensitivity. The androgen receptor is a ligand-dependent transcription factor that has a polymorphism involving CAG (glutamine) trinucleotide repeats. The longer the length of the CAG repeat, the less the transactivation activity of the ligand-bound receptor. In a study of 35 patients with Klinefelter syndrome, the one genetic factor affecting phenotype was the CAG repeat length of the functional androgen receptor. The repeat lengths correlated inversely with penile length, a T-sensitive organ. Neither an effect of parental origin of the X chromosome (imprinting) nor evidence of skewed X chromosome inactivation affected the phenotype. In a study of 77 patients with Klinefelter syndrome, preferential inactivation of the shorter CAG allele was detected.
The most consistent feature of Klinefelter syndrome is small atrophic testes due to seminiferous tubule dysgenesis and consequent azoospermia. Tall stature is common in Klinefelter syndrome, reflecting the presence of three SHOX genes. Adult males with a 47, XXY karyotype tend to be taller than average, with adult height close to the 75th percentile, mainly because of the disproportionate length of their legs, which is present prepubertally. Untreated adult males, especially those with subnormal sex steroid levels, are at increased risk of osteoporosis. In addition, individuals with Klinefelter syndrome have an increased prevalence of mild diabetes mellitus, autoimmune thyroid disorders, early tooth decay, varicose veins, stasis dermatitis, cerebrovascular disease, chronic pulmonary disease, deep vein thrombosis, carcinoma of the breast, midline germ cell tumors, and Hodgkin lymphoma. The data on carcinoma of the breast are controversial. There were no cases in a cohort of 696 men with Klinefelter syndrome from the Danish cytogenetic register; however, 7 of 93 men with breast cancer in a Swedish study had Klinefelter syndrome. Life expectancy is reduced by about 2.1 years over controls, with a lower death rate than expected from ischemic heart disease and prostate cancer.
Prepubertally, Klinefelter syndrome is characterized by small undescended testes (> 25% of patients), relatively reduced penile length, clinodactyly, hypotonia, disproportionately long legs, personality and behavioral disorders, and a lower mean verbal IQ score. There is no significant difference in full-scale IQ. Deficits are found primarily in specific areas of cognition such as language and problem solving. Severe mental retardation requiring special schooling is uncommon. Gynecomastia and signs of androgen deficiency such as diminished facial and body hair, a small phallus, and poor muscular development occur postpubertally in affected patients.
Patients with Klinefelter syndrome may have delayed adolescence although a recent study suggested that the onset of puberty and early progression were relatively normal. There is an increased risk of malignant extragonadal germ cell tumors in the central nervous system, mediastinum, and sacral region. The germ cell tumors often secrete hCG and cause sexual precocity in the affected prepubertal individual. Accordingly, we recommend that boys with hCG-secreting midline germ cell tumors have a karyotype analysis.
The testicular lesion is progressive and gonadotropin dependent. Gonadal germ cells become depleted at an accelerated rate after the onset of puberty, and there appears to be a maturation arrest of spermatogenesis. The testes are characterized in the adult by extensive seminiferous tubular hyalinization and fibrosis, absent or severely deficient spermatogenesis, and pseudoadenomatous clumping of the Leydig cells. Although hyalinization of the tubules is usually extensive, it varies considerably from patient to patient and even between testes in the same patient. Azoospermia is the rule; patients reported to be spontaneously fertile invariably have been 46,XY/47,XXY mosaics. Using surgical techniques, sperm has been found in 50% of the nonmosaic patients with Klinefelter syndrome. In biopsied testes of patients with 47,XXY Klinefelter syndrome, only a minor fraction of tubules contain germ cells. FISH analysis revealed that the spermatocytes were euploid, suggesting that the spermatogonia that undergo mitosis have only one X chromosome thus allowing for progression in cell differentiation and the ability to engage in meiosis. Intracytoplasmic sperm injection (ICSI) with sperm retrieved surgically from testes (TESE) of patients with Klinefelter syndrome has been used with success for ovum fertilization. Using ICSI, most infants born to fathers with Klinefelter syndrome have had normal karyotypes. However, there is an increased incidence of sex chromosome and autosomal aneuploidy, possibly related to the assisted fertility technique (ART). Preimplantation genetic diagnosis (PGD) is recommended. However, the potential risk to the embryo of PGD must be weighed against the risk of chorionic villus biopsy or amniocentesis in the event of a successful pregnancy.
Nondisjunction during the first or second meiotic division of gametogenesis plays an important role in the genesis of a 47,XXY karyotype. Fifty-three percent of cases appear to result from paternal nondisjunction at the first meiotic division, 34% from meiotic nondisjunction during the first maternal meiotic division, and 9% from nondisjunction at the second meiotic division. Only 3% of patients appear to have arisen from postzygotic mitotic nondisjunction. Early studies suggested that parental age was a factor in the etiology of Klinefelter syndrome, but in a study of 228 patients, no association with advanced parental age was found.
Diagnosis: Klinefelter syndrome is suggested by the classic phenotype and hormonal changes. It is confirmed by the finding of an X chromatin-positive buccal smear or an FISH analysis which demonstrates an XXY constitution or the demonstration of a 47,XXY karyotype in blood, skin, or gonads. After puberty, levels of serum gonadotropins especially follicle-stimulating hormone (FSH) are raised. The T production rate, the total and free levels of T, and the metabolic clearance rates of T and estradiol tend to be low, whereas plasma estradiol levels are relatively normal or high for a male. Testicular biopsy reveals the classic findings of hyalinization of the seminiferous tubules, severe deficiency of spermatogonia, and pseudoadenomatous clumping of Leydig cells.
Hormonal evaluation in the newly diagnosed patient with Klinefelter syndrome should include plasma LH and FSH by immunochemiluminescence (ICMA). These levels are usually in the normal range prior to the onset of puberty. Thereafter, a rise in FSH and subsequently LH is usually observed. A plasma estradiol and T by liquid chromatography-tandem mass spectrometry (LCMSMS) should be obtained. In the adolescent or postadolescent patient, a bone density is indicated to determine whether the patient has osteopenia/osteoporosis and requires therapy for this.
Treatment: Treatment in adolescents and adults with Klinefelter syndrome should be directed toward health maintenance and the options for fertility (eg, cryopreservation of sperm from ejaculate in adolescence or TESE in older patients, T replacement if necessary, and prevention of osteopenia/osteoporosis and deep vein thrombosis). Klinefelter patients are born with spermatogonia, and testicular failure is progressive beginning at puberty. Obtaining ejaculated sperm in early adolescence for cryopreservation, if possible, might obviate the need for TESE at a later time. Although several studies have reported that sperm have been found in 8% of ejaculated serum sample fron nonmosaic 47,XXY patients, a recent study reported lack of sperm in samples from 13 patients less than 20 years of age. Because of the progressive nature of testicular failure which occurs after the onset of puberty, surgical sperm retrieval is less successful with advancing age. T therapy in the hypogonadal patient will enhance secondary sexual characteristics and sexual performance, increase muscle mass, prevent or help ameliorate osteopenia/osteoporosis and possibly prevent the development of gynecomastia. If T deficiency is evident in adolescence, therapy with T enanthate in oil at a dose of 50 mg intramuscularly monthly may be initiated. Alternatively, testosterone by patch or gel may be used. The plasma concentration of T can be assessed midway between injections, and the doses gradually increased to attain a level in the mid-normal range for stage of development and/or age.
In the adult patient, transdermal T by patch or gel can be used for replacement therapy. T levels can be assessed 1 week after the start of therapy and adjusted to maintain levels in the mid-normal range for age. Patients should be informed of the possibility of deep vein thrombosis and the T discontinued if the hematocrit rises above 54% because of the risk of hypercoagulability associated with elevated hematocrit levels. If gynecomastia is present and is not ameliorated by T therapy, surgical reduction is required if it is psychologically disturbing to the patient.
Early diagnosis with remedial therapy for speech and behavior problems, counseling for the patient and parents, as well as support group services appears to improve the well being and overall quality of life in patients diagnosed with Klinefelter syndrome.
Variants of Klinefelter syndrome include 46,XY/47,XXY mosaics as well as patients with multiple X and Y chromosomes. With increasing numbers of X chromosomes in the genome, both mental retardation and other developmental anomalies such as radioulnar synostosis become prevalent.
Phenotypic males with a 46,XX karyotype have been described since 1964; the incidence of 46,XX males is approximately 1:20,000 births. In general, these individuals have a male phenotype, male psychosocial gender identity, and testes with histologic features similar to those observed in patients with a 47,XXY karyotype. At least 10% of patients have hypospadias or ambiguous external genitalia, primarily those patients who are SRY negative. 46,XX males have normal body proportions and a mean final height that is shorter than that of patients with a 47,XXY sex chromosome constitution or normal males but taller than that of normal females. There is a higher incidence of undescended testes in 46,XX males than in 47,XXY patients. As in 47,XXY individuals, T levels are low or low-normal, gonadotropins are elevated, and spermatogenesis is impaired postpubertally. Gynecomastia is more frequent than that observed in 47,XXY males.
Males with a 46,XX karyotype have acquired one X chromosome from each of their parents. Approximately 90% of XX males have a Y chromosome–specific DNA segment from the distal portion of the Y short arm translocated to the distal portion of the short arm of the paternal X chromosome. This translocated segment is variable in length but always includes the SRY gene, which encodes testis-determining factor as well as the pseudoautosomal region of the Y chromosome. Thus, in at least 90% of XX males, an abnormal X-Y terminal exchange during paternal meiosis has resulted in two products: an X chromosome with an SRY gene and a Y chromosome deficient in this gene (the latter would result in a female with XY gonadal dysgenesis). Fewer than 10% of XX males tested have lacked Y chromosome–specific DNA sequences (including the SRY gene) and the pseudoautosomal region of the Y chromosome. These XX, SRY-negative males tend to have hypospadias and may have relatives with ovotesticular DSD.
The finding of XX males who lack any evidence of Y chromosome–specific genes suggests that testicular determination—and, thus, male differentiation—can occur in the absence of a gene or genes from the Y chromosome. This could be a result of (1) mutation (upregulation) or duplication of a downstream autosomal gene involved in male sex determination (eg, SOX9 which if duplicated in a 46,XX individual causes sex reversal both in mice and humans); or (2) mutation, deletion, or aberrant inactivation of a gene sequence, which causes repression of testis determination. Two families with multiple 46,XX phenotypically normal males who had palmoplantar hyperkeratosis and a predisposition to squamous cell carcinomas have been reported. All affected males had a homozygous deletion of 2700 base pairs of the R-spondin1 gene. RSPO1, an ovary-determining gene, has no role in testes determination as evidenced by the description of a 46,XY male with a homozygous mutation of RSPO1 who had normal testicular function; or (3) circumscribed Y chromosome mosaicism (eg, occurring only in the gonads); (4) A 46,XX SRY negative male has recently been described with a mutation in SOX3. SOX3 is a member of the SRY HMG box family of genes and is found on the X chromosome at Xp26-27. This mutation is postulated to cause aberrant expression of SOX3 in gonadal primordia and thereby upregulating SOX9 expression resulting in testicular determination.
Syndrome of Gonadal Dysgenesis: Turner Syndrome and Its Variants
One in 2000 to 2500 newborn females have an absence of a second sex chromosome (either an X or Y), a structural rearrangement of the X chromosome, and/or mosaicism. Patients with a 45,X karyotype represent approximately 50% of all patients with X chromosome abnormalities. It has been estimated that 99% of 45,X fetuses do not survive beyond 28 weeks of gestation probably related to congenital heart anomalies, and 15% of all first-trimester abortuses have a 45,X karyotype. In about 70% to 80% of 45,X individuals, the X chromosome is of maternal origin.
The cardinal features of 45,X gonadal dysgenesis are a variety of somatic anomalies, sexual infantilism at puberty secondary to gonadal dysgenesis, and short stature. Patients with a 45,X karyotype can be recognized in infancy, usually because of lymphedema of the extremities and loose skin folds over the nape of the neck. In later life, the typical patient is often recognizable by her distinctive facies in which micrognathia, epicanthal folds, prominent low-set ears, a fish-like mouth, and ptosis are present to varying degrees. The chest is shield-like, and the neck is short, broad, and webbed (40% of patients). Additional anomalies associated with Turner syndrome include renal abnormalities (30%), pigmented nevi, cubitus valgus, congenital hip subluxation, a tendency to keloid formation, lymphedema of the lower extremities or puffiness of the dorsum of the hands and feet, short fourth metacarpals and metatarsals, Madelung deformity of the wrist, scoliosis, and recurrent otitis media, which may lead to conductive hearing loss. The high prevalence of otitis media is a consequence of an abnormal relationship between the Eustachian tube and the middle ear. Sensorineural hearing loss increases with age and is present in 61% of women with Turner syndrome over 35 years of age. All patients should have routine periodic auditory screening.
Patients with Turner syndrome have an increased prevalence of cardiovascular anomalies. The incidence of bicuspid aortic valves diagnosed by echocardiography has varied between 9% and 34%. A bicuspid aortic valve is a risk factor for subacute bacterial endocarditis. With age there is an increased tendency for a stenotic and/or insufficient aortic valve. Coarctation of the aorta is present in 10% of patients, especially individuals with a webbed neck. Bicuspid aortic valves, coarctation, aortic dilation, and hypertension are critical risk factors for aneurysm formation and death due to aortic rupture. Thus, all patients with Turner syndrome should have a complete cardiac evaluation by a cardiologist at diagnosis as well as periodic follow-up echocardiography or magnetic resonance imaging (MRI) as necessary to monitor aortic diameter and cardiovascular status. Routine intravenous urography or renal sonography is indicated to detect a surgically correctable renal abnormality. The most common renal anomalies are a horseshoe kidney, duplication of the renal pelvis and ureter, and hydronephrosis secondary to uteropelvic obstruction. Complete absence of a kidney and gross renal ectopia have been reported. The internal ducts as well as the external genitalia of these patients are invariably female except in rare patients with a 45,X karyotype, in whom a Y-to-autosome or Y-to-X chromosome translocation or cryptic mosaicism for a Y-containing cell line has been found.
Short stature is an invariable feature of the syndrome of gonadal dysgenesis. Mean final height in 45,X patients is 143 cm, with a range of 133 to 153 cm. Short stature found in patients with the syndrome of gonadal dysgenesis is not due to a deficiency of growth hormone (GH), insulin-like growth factor I (IGF-I), sex steroids, or thyroid hormone. It is related, at least in part, to haploinsufficiency of the SHOX gene in the pseudoautosomal region of the X and Y chromosome (see earlier sections). The administration of high-dose recombinant human GH leads to an increase in final height.
Gonadal dysgenesis is a cardinal feature of patients with a 45,X chromosome constitution. The gonads at adolescence are typically streak-like and usually contain only fibrous stroma arranged in whorls. Longitudinal studies of both basal and gonadotropin-releasing hormone (GnRH)-evoked gonadotropin secretion in patients with gonadal dysgenesis indicate a lack of feedback inhibition of the hypothalamic-pituitary axis by the dysgenetic gonads in affected infants and adolescents (Figure 14–12). Plasma and urinary gonadotropin levels, particularly FSH levels, are high during early infancy and after 9 to 10 years of age. Because ovarian function is impaired, puberty does not usually ensue spontaneously; sexual infantilism is a hallmark of this syndrome. Rarely, patients with a 45,X karyotype may undergo spontaneous pubertal maturation, menarche, and even pregnancy. It has been suggested that measurable levels of inhibin B may indicate the presence of functional follicles and the possibility of spontaneous pubertal development while low levels of AMH might herald ovarian failure. A variety of other disorders are associated with this syndrome, including obesity, nonosteoporotic fractures in childhood and osteoporotic fractures in adulthood, congenital dislocation of the hip, type 2 diabetes mellitus, an increased number of melanocytic nevi but no increase in melanoma, orthodontic problems, Hashimoto thyroiditis, rheumatoid arthritis, inflammatory bowel disease, intestinal telangiectasia with bleeding, hypertension, coeliac disease, ischemic heart disease, stroke, and anorexia nervosa. Abnormal liver enzymes with elevation of transaminases, alkaline phosphatase, and gamma-glutaryl transferase have been reported in more than 50% of adults. Most patients with Turner syndrome have normal intelligence. Only a small percentage of patients have significant developmental delay. However, 70% have learning disabilities involving spatial relationships, nonverbal problem-solving, and attention. Early testing and remediation is recommended, as well as psychologic support.
Figure 14–12
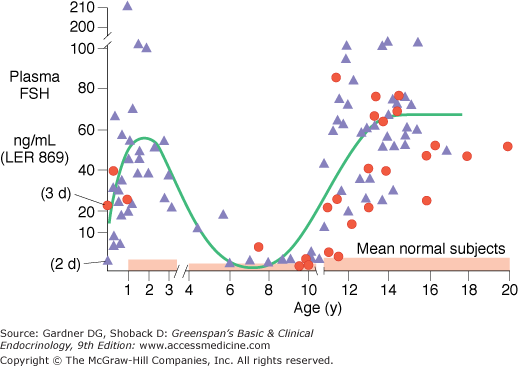
Diphasic variation in basal levels of plasma follicle-stimulating hormone (FSH) (ng/mL-LER 869) in patients with a 45,X karyotype (solid triangles) and patients with structural abnormalities of the X chromosome and mosaics (solid circles). Note that mean basal levels of plasma FSH in patients with gonadal dysgenesis are in the castrate range before 4 years and after 10 years of age. (Reproduced with permission from Conte FA, Grumbach MM, Kaplan SL. A diphasic pattern of gonadotropin secretion in patients with the syndrome of gonadal dysgenesis. J Clin Endocrinol Metab. 1975;40:670.)
Diagnosis: Phenotypic females with the following features should have a karyotype analysis: (1) short stature (>2.5 standard deviations [SDs] below the mean value for age) with or without somatic anomalies associated with the syndrome of gonadal dysgenesis; and (2) delayed adolescence with an increased concentration of plasma FSH; (3) patients with clinical findings suggestive of Turner syndrome (eg, lymphedema of hands or feet, left-sided cardiac anomalies, characteristic facies, short fourth metacarpals, etc).
The initial evaluation of a patient with Turner syndrome should include karyotype analysis as well as FISH of 200 cells to rule out mosaicism or the presence of a Y chromosome in individuals with a 45,X karyotype and in those in whom a small nonidentifiable marker chromosome is present. A renal ultrasound is indicated to rule out surgically correctable anomalies. A cardiac evaluation with echocardiogram or MRI should be done to evaluate aortic size and rule out coarctation of the aorta and an anomaly of the aortic valve. Besides routine chemistries, gonadotropins, AMH (provides an estimate of the number of antral follicles), and inhibin B should be assessed in order to determine the status of the gonads. Periodic assessment of the cardiac status, especially in those with bicuspid aortic valves and/or hypertension, is indicated throughout the lifetime of the patient. Other systems which need periodic evaluation include: (1) thyroid function studies, (2) assessment of tissue transglutaminase activity for celiac disease, (3) hearing evaluation, (4) orthodontics, and (5) psychosocial and school performance issues. In view of the increased incidence of specific learning defects involving spatial relationships, executive skills, and so forth, it is prudent to evaluate these functions at 4 to 5 years of age so that appropriate remedial therapy can be undertaken as early as possible if warranted.
At adolescence, in addition to the continuing above assessments, lipids, fasting glucose and insulin, and liver function should be assessed. Psychosocial and educational issues should continue to be addressed with the patient and the parents.
Treatment: Maximizing growth and hormone replacement at puberty in order to engender an orderly progressive development of secondary characteristics at a time concordant with peers are two of the main goals of therapy in patients with Turner syndrome.
Individuals with Turner syndrome treated with recombinant GH (0.375 mg/kg/wk divided into seven once-daily doses (maximum dose 15-18 mg/wk), with or without oxandrolone (0.0625 mg/kg/d by mouth), have an increase in the rate of growth that is sustained and results in a mean increase in height of 7 to 10 cm after 3 to 7 years of therapy. The serum IGF-1 level should be followed and the dose titered to maintain the IGF-1 level below the upper limit of normal for age. Beginning GH therapy earlier (as soon as growth failure is evident) in childhood allows for a longer duration of therapy which results in a greater gain in height and fosters initiation of estrogen replacement at an age commensurate with normal puberty. Before initiation of GH therapy, a thorough analysis of the costs, benefits, and possible side-effects must be discussed with the parents and the child. Significant side effects include: (1) the accelerated onset of diabetes mellitus in the prediabetic, (2) pseudotumor cerebri, (3) slipped femoral epiphysis, and (4) unknown long-term side effects. The influence of the gain in height on any of the quality of life parameters in individuals with Turner syndrome treated with GH has been questioned by some. There is no synergistic effect of combined estrogen replacement and GH therapy on final height in this disorder. However, in a recent study, percutaneous estrogen administration for pubertal induction was associated with a 2.1 cm greater adult height than oral estrogen administration. These data suggest that, as observed in adult women, oral estrogens decrease the plasma concentrations of IGF-1 and suppress IGF-1’s metabolic effects. In patients who have been treated with GH and have achieved an acceptable height and in those who have refused GH therapy and have evidence of gonadal dysgenesis, estrogen replacement therapy is usually initiated after 11 to 13 years of age.
Increasingly, transcutaneous estrogen administration by patch or gel, which avoids first pass through the liver, is the treatment of choice to induce female secondary sexual characteristics in hypogonadal girls. In a study of 15 patients, an estradiol patch was placed nocturnally for 12 hours to simulate the diurnal pattern of estradiol secretion in early puberty. By cutting the 25-μg patch (Vivelle-Dot) when necessary and measuring serum estradiol concentrations, it was concluded that 0.08 to 0.12 μg/kg of body weight was an appropriate starting dose of estradiol transcutaneously. Plasma concentrations of estradiol were measured in the morning and maintained in a range appropriate for Tanner stage by adjusting the dose. The starting dose was usually maintained for 9 months before increasing the dose. The aim was to achieve midpuberty estradiol levels by 15 months. A progestin was added to the estradiol within 2 years after the start of estradiol.
Conjugated estrogens (0.3 mg or less) or ethinyl estradiol (3-5 μg) can be given orally for the first 21 days of the calendar month. Thereafter, the dose of estrogen is gradually increased over the next several years to 1.25 mg of conjugated estrogens or 10 μg of ethinyl estradiol daily for the first 21 days of the month. The minimum dose of estrogen necessary to initiate and maximize uterine development, secondary sexual characteristics, and menses, as well as to prevent osteoporosis should be administered. After the first 18 to 24 months of estrogen therapy, medroxyprogesterone acetate (5 mg) or a comparable progestin is given on the tenth to twenty-first days of the month to ensure physiologic menses and to reduce the risk of endometrial carcinoma, which is associated with unopposed estrogen stimulation.
We recommend a transition program when transferring adolescent or young adult patients to an adult caregiver. Ideally, this program will introduce the patient to the physicians who will be involved in her future care which may include an internist, endocrinologist, reproductive endocrinologist, ENT specialist, adult cardiologist, and psychologist.
Many of the morbidities that contribute to the decrease in lifespan are detectable with comprehensive medical surveillance and are amenable to medical and/or surgical therapy. Protocols for the management of the adult Turner syndrome patient have been published (see Gravholt et al; Bondy et al; Conway in reference list at the end of this chapter). Individuals with Turner syndrome by nature are very maternal. They should be reassured about their sexual function and informed about their potential for maternity with donor eggs and in vitro fertilization in an age-appropriate manner. The success rate of ovum donation is 40% per treatment cycle, and only 50% of pregnancies achieve a live birth due to a high miscarriage rate. In one study, 38% of patients had pregnancy associated hypertensive problems. There is an increased risk of aortic dissections during pregnancy. Thus, all women with Turner syndrome who desire a pregnancy should have a careful, complete cardiac evaluation prior to egg implantation and close continued monitoring if pregnancy ensues. We recommend caesarian delivery to avoid the increased stress to the cardiovascular system associated with labor and because of the risk of cephalopelvic dystocia in these women.
Patients with structural abnormalities of the X chromosome (deletions and additions) and sex chromosome mosaicism with a 45,X cell line may manifest the somatic as well as the gonadal features of the syndrome of gonadal dysgenesis . Evidence suggests that genes on both the long and short arms of the X chromosome control gonadal differentiation, whereas genes primarily on the short arms of the X prevent the short stature and somatic anomalies that are seen in 45,X patients (see Figure 14–2). In general, 45,X/46,XX mosaicism modifies the 45,X phenotype toward normal and can even result in normal gonadal function and fertility. Patients with duplication of the long arms of the X chromosome and deletion of the short arm—the so-called Xq isochromosome—appear to have an increased prevalence of autoimmune thyroiditis, type 2 diabetes, and inflammatory bowel disease in comparison to those with a 45,X karyotype. Patients with a 46,XrX (ring) cell line in their karyotype may manifest mental retardation and congenital anomalies not usually associated with Turner syndrome. These abnormalities are related to lack of inactivation of small ring X chromosomes and, hence, functional disomy for genes on the ring X chromosome and the normal X chromosome.
These patients usually have mosaicism with a 45,X and a Y-bearing cell line—45,X/46,XY; 45,X/47,XXY; 45,X/46,XY/47,XYY—or perhaps a structurally abnormal Y chromosome. They range from phenotypic females with the features of Turner syndrome to patients with ambiguous genitalia to completely normal-appearing males with few stigmas of Turner syndrome and short stature. The variations in gonadal differentiation range from bilateral streaks to bilateral dysgenetic testes to apparently normal testes, and there may be asymmetric development (eg, a streak on one side and a dysgenetic testis or, rarely, a normal testis on the other side)—sometimes called mixed gonadal dysgenesis. The development of the external genitalia and of the internal ducts correlates with the degree of testicular differentiation and the capacity of the fetal testes to secrete AMH and T.
The risk of gonadal tumors is greatly increased in patients with 45,X/46,XY mosaicism and streak or dysgenetic gonads; hence, prophylactic removal of streak gonads or dysgenetic undescended testes in this syndrome is indicated. Breast development at or after the age of puberty in these patients is commonly associated with a gonadal neoplasm, usually a gonadoblastoma. Pelvic sonography, computed tomography (CT), or MRI may be useful in screening for neoplasms in these patients. Gonadoblastomas are calcified and so may be visible even on a plain film of the abdomen.
Diagnosis: The diagnosis of 45,X/46,XY mosaicism can be established by the demonstration of both 45,X and 46,XY cells in blood, skin, or gonadal tissue. In some mosaics, a marker chromosome is found that is cytogenetically indistinguishable as X or Y. In these cases, either FISH or molecular analyses with X- and Y-specific probes is indicated to definitively determine the origin of the marker chromosome, because gonadoblastomas have been reported in patients with deleted Y chromosomes—even those with deletions of the SRY gene. Ninety percent of patients with 45,X/46,XY mosaicism ascertained by amniocentesis have normal male genitalia and normal testicular histology. Thus, the ambiguity of the genitalia invariably described in patients with 45,X/46,XY mosaicism is due to ascertainment bias. We have observed a short 30-year-old male with documented 45,X/46,XY mosaicism who has normal male genitalia and is fertile.
Treatment:
Related posts:
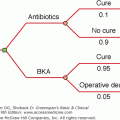


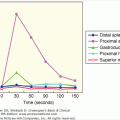
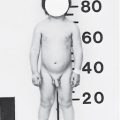
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


