Chapter 19
Disorders of Mitochondrial Energy Metabolism, Lipid Metabolism and Other Disorders
Marjorie Dixon, Jacky Stafford, Fiona White, Nicol Clayton and Janine Gallagher
An Introduction to Inherited Metabolic Disorders may be found at the beginning of Chapter 17.
Disorders of Mitochondrial Fatty Acid Oxidation and Related Disorders
Marjorie Dixon
Introduction to mitochondrial fatty acid oxidation
Mitochondrial fatty acid β-oxidation is the primary breakdown pathway of fatty acids to generate energy. Fatty acids are a major fuel source for most tissues of the body, especially during fasting, when glucose supply is limited. They are the principal energy source for cardiac muscle and skeletal muscle in the resting state and during prolonged exercise. Fatty acid oxidation is also important for the production of ketone bodies; these are another important fuel which can be used by all tissues and particularly by the brain during prolonged fasting as it cannot use fatty acids to generate energy.
Fatty acids consist of a hydrocarbon chain with a carboxylic acid group at one end. Most naturally occurring fatty acids have a chain length of 16–18 carbon atoms and are referred to as long chain fatty acids. Fatty acids are stored in adipose tissue as triglycerides, in which the carboxylic acid groups of three fatty acids are esterified to glycerol. Fat metabolism begins in adipose tissue as a response to falling levels of blood glucose. Insulin activates hormone-sensitive lipase which promotes the release of free fatty acids from triglycerides into the blood stream. The fatty acids are then transported to the tissues bound to albumin. In tissues where they are used, fatty acids enter the mitochondria bound to carnitine. This pathway is called the carnitine cycle or shuttle.
Carnitine cycle/shuttle
The carnitine cycle involves three enzymes which transport long chain fatty acids into the mitochondria. The first step occurs at the outer mitochondrial membrane where long chain fatty acids are activated to a coenzyme A ester, then to an acylcarnitine by the action of carnitine palmitoyl transferase I (CPTI); next it is transported by carnitine acylcarnitine translocase (CACT) into the mitochondria where it is converted back to an acyl-CoA ester by carnitine palmitoyl transferase II (CPTII), ready to undergo mitochondrial β-oxidation (Fig. 19.1). Carnitine is required in this process; it is transported inside the cells by an organic cation transporter (OCTN2) present in the heart, muscle and kidney. CPTI is the regulatory step for fatty acid oxidation. Malonyl CoA is involved in the regulation of CPTI, so indirectly controls fatty acid oxidation [1]. Circulating levels of malonyl CoA are high during the fed state and inhibit CPTI; conversely in the fasting state they fall and activate CPTI.
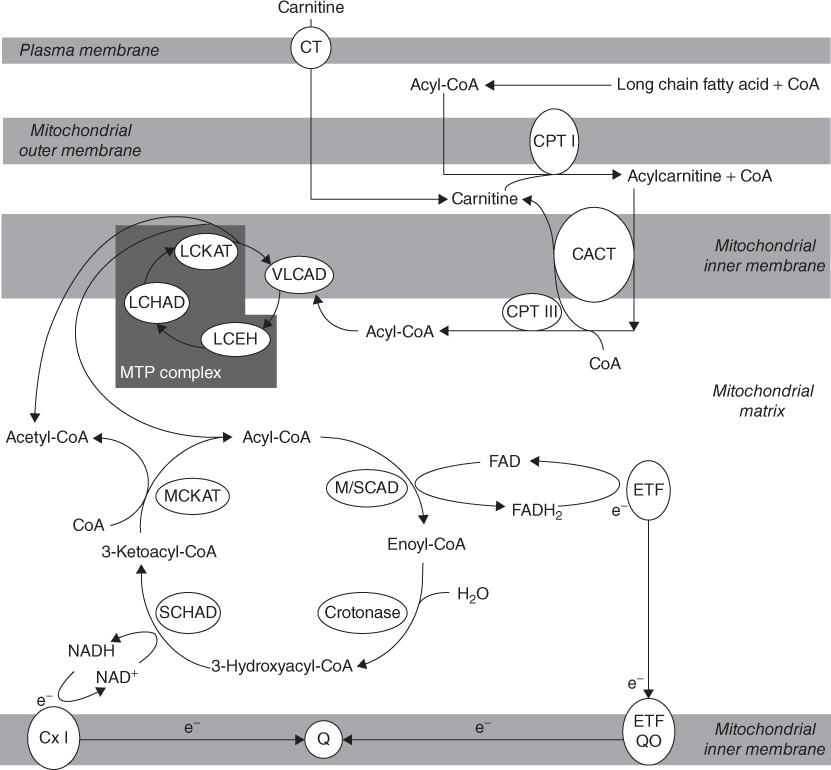
Figure 19.1 Biochemical pathways of mitochondrial fatty acid oxidation (permission to reproduce) Enzymes involved: CT, carnitine transporter; CPTI, carnitine palmitoyltransferase I; CACT, carnitine acylcarnitine translocase; CPTII, carnitine palmitoyl transferase; VLCAD, very long chain acyl-CoA dehydrogenase; LCEH, long chain enoyl Co-A hydratase; LCHAD, long chain 3-hydroxyacyl-CoA dehydrogenase; LCKAT, long chain ketoacyl-CoA thiolase; MTP, mitochondrial trifunctional protein; M/SCAD, medium or short chain acyl-CoA dehydrogenase; SCHAD, short chain acyl-CoA dehydrogenase; MCKAT, medium chain ketoacyl-CoA thiolase; CxI, complex I of respiratory chain; Q, ubiquinone; e−, electrons; ETF, electron transfer flavoprotein; ETFQO, electron transfer flavoprotein ubiquinone oxidoreductase.
Reprinted with permission of Springer.
Mitochondrial β-oxidation
Within mitochondria, fatty acids are broken down by the spiral pathway of β-oxidation (Fig. 19.1). Every turn of the spiral involves four steps, catalysed by several enzymes of different chain length specificities. The first step, for example, is catalysed by very long chain, medium chain and short chain acyl-CoA dehydrogenases (VLCAD, MCAD, SCAD). The third step in the β-oxidation spiral is another dehydrogenation reaction and is catalysed by long and short chain 3-hydroxyacyl-CoA dehydrogenases (LCHAD, SCHAD). Both of these dehydrogenation reactions release energy which can be harnessed for use by the cell. This is achieved by passing electrons to the mitochondrial respiratory chain, either directly or via electron transfer flavoprotein (ETF) and ETF ubiquinone oxidoreductase (ETFQO). In addition to producing energy, each turn of the β-oxidation spiral releases a molecule of acetyl-CoA and shortens the fatty acid by two carbon atoms. Acetyl-CoA can be metabolised in the tricarboxylic acid cycle, but in the liver it is converted to ketone bodies via 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) synthase and lyase. A more detailed description of the biochemical basis of mitochondrial fatty acid oxidation is described elsewhere [2, 3].
Overview of disorders of mitochondrial fatty acid oxidation, ketogenesis and ketolysis
A number of disorders of the mitochondrial fatty acid oxidation pathway have now been described. These vary in severity and may present at any time from the neonatal period to adulthood [2, 3]. The clinical features associated with fatty acid oxidation disorders (FAOD) probably result from the failure of the production of energy and accumulation of toxic intermediates [4]. The commonest disorder is medium chain acyl-CoA dehydrogenase deficiency (MCADD). Typically, this presents in early childhood with hypoglycaemia and acute encephalopathy brought on by fasting or an acute infection. The fatty acid oxidation defect impairs the production of ketone bodies and the hypoglycaemia is therefore ‘hypoketotic’. Most other defects affect long chain fatty acid oxidation, either because they involve long chain specific β-oxidation enzymes, e.g. very long chain acyl-CoA dehydrogenase deficiency, or because they affect the carnitine mediated entry of long chain fatty acids into mitochondria, e.g. carnitine palmitoyl transferase deficiency type I. Defects of ETF and ETFQO cause multiple acyl-CoA dehydrogenase deficiency (MADD); in this condition there is impaired oxidation of long chain, medium chain and short chain fatty acids and various other substrates. Patients with long chain fatty acid oxidation defects may present with hypoglycaemia and encephalopathy, as in MCADD. They may often have additional problems such as cardiomyopathy, weakness or episodes of muscle breakdown (rhabdomyolysis).
The synthesis of ketone bodies occurs via the intermediate HMG-CoA and depends primarily on the enzymes HMG-CoA synthase and HMG-CoA lyase. Deficiencies of these enzymes are known as the disorders of ketogenesis and present with ‘hypoketotic’ hypoglycaemia. There are also defects in the utilisation of ketone bodies; the enzymes involved are succinyl-CoA 3-oxoacid-CoA transferase (SCOT) and 2-methylacetoacetyl-CoA thiolase (MAT). Deficiencies of these enzymes are known as the disorders of ketolysis or ketone body utilisation defects. They present primarily with a severe ketoacidosis.
Disorders of fatty acid oxidation and ketone bodies are treated primarily by diet. Some aspects of management are universal but others vary with the underlying disorder, age at onset and severity. The main aim is to minimise the oxidation of fatty acids by using an emergency regimen during illness and avoiding fasting. Additionally in severe defects of long chain fatty acid oxidation the intake of dietary fat is limited and medium chain fats may be used.
Medium chain acyl-CoA dehydrogenase deficiency
MCADD is the commonest of the fatty acid oxidation defects, with an incidence of 1 in 10 000 to 27 000 in babies of Northern European descent. MCADD is due to a deficiency of the enzyme medium chain acyl-CoA dehydrogenase which is necessary for the oxidation of medium chain fatty acids (carbon chain length C6–C12). Ketone body and energy production is reduced and there is accumulation of medium chain fatty acids, specifically octanoyl carnitine (C8), which are considered to be responsible for the clinical sequelae. Clinically, MCADD usually presents between 6 months and 4 years of age but neonatal and adult onset can also occur. Some patients remain asymptomatic throughout life. The typical picture is of encephalopathy with ‘hypoketotic’ hypoglycaemia, precipitated by metabolic stress such as fasting, gastrointestinal illness or respiratory infections [5–7]. Alcohol intoxication is another precipitating cause seen in adult patients [8]. Without prompt treatment there is a high morbidity and mortality; some patients die suddenly, probably due to cardiac arrhythmias [5–7]. Between episodes patients are usually completely well because, under basal conditions, oxidation of medium chain fatty acids is near normal [9]. Once diagnosed the outlook is much improved [10].
Genetics
MCADD is inherited as an autosomal recessive disorder, so a baby has a 1 in 4 chance of inheriting the disorder from its parents. Due to a founder effect the c.985A>G mutation is common in Northern Europeans and is associated with classical clinical disease. Screening has resulted in the identification of further mutations, some of which have never been reported in clinically presenting patients [11]. Currently, in the UK all babies are treated irrespective of mutation.
Expanded newborn screening for MCADD
MCADD is part of the existing blood spot newborn screening programme in the UK (since 2009 for England, all of the UK since 2012). In England newborn screening identifies about 60 babies per year, with a prevalence of about 1 in 10 000 [12]. For this a heel prick blood sample is collected between 5 and 8 days of age on the standard newborn screening collection cards. MCADD screening uses the quantitative assay of C8 and decanoylcarnitine (C10). More details on screening, diagnostic and clinical management protocols in the UK can be found at www.newbornbloodspot.screening.nhs.uk/mcadd and www.bimdg.org.
Dietary management
At risk neonate with a family history of MCADD
Neonatal deaths have been reported in MCADD in breast and formula fed babies [13, 14]. Newborns are at greatest risk during the first 72 hours of life, especially if they are being breast fed (see below). Therefore it is essential that at risk infants who have a family history of MCADD receive adequate feeds during the first few days of life. The 2012 UK guidelines for managing at risk babies for MCADD can be accessed from the above websites and are as follows:
- any at risk babies should be screened at 24–48 hours and results made available, promptly
- the baby should be fed regularly, a term baby at least 4 hourly and preterm 3 hourly day and night until a diagnosis of MCADD is confirmed or excluded
- breast fed babies are at particular risk because the energy content of breast milk is low for the first few days and only small volumes of breast milk are available and initially consumed; top-ups of formula milk are therefore recommended
- a volume of 60 mL/kg/day of standard infant formula is advised for the first three complete days (72 hours); ideally this should be divided evenly between 6 to 8 feeds and given after the breast feed
- if there are concerns about the baby or they are taking inadequate volumes of feeds they should be transferred to the neonatal unit and fed by nasogastric tube or given an intravenous infusion containing 10% glucose
Dietary management overview
Normal diets contain predominantly fatty acids with a chain length of C16 and C18. These long chain fatty acids undergo β-oxidation to release energy and the carbon chain length is progressively shortened to a medium, then short chain length. Under basal conditions, the oxidation of medium chain fatty acids has been reported to be near normal in patients with MCADD due to overlapping enzyme substrate specificity [9]. This only becomes a problem when there is an increased demand for fatty acids to provide energy such as during fasting or illness. The well child can therefore have a normal diet without restriction of long chain fat. It is important, however, to avoid prolonged fasts as fatty acid oxidation rates increase with fasting. In infants, fatty acid oxidation rates rise after a shorter duration of fasting and frequent feeding is therefore recommended. There are few published data on the safe duration of fasting in MCADD. Derks et al. have suggested treatment guidelines based on data from the literature and Dutch MCADD patients, but they note the paucity of data in the first 6 months of life [15]. Compared with the UK guidelines (Table 19.1) the fasting times are more restricted in younger children. Illness in any child is usually associated with loss of appetite and prolonged fasting; fatty acid oxidation rates increase to provide essential energy sources. In MCADD this process is limited and the child is at risk of encephalopathy. To prevent encephalopathy an emergency regimen (ER) of frequent feeding of high carbohydrate (CHO) drinks/feeds needs to be given. This will provide energy, stimulate insulin secretion and thereby minimise lipolysis and fatty acid oxidation.
Table 19.1 Medium chain acyl-CoA dehydrogenase (MCAD) deficiency: guidelines for ‘maximum safe fasting times’ for the well child
| Age | Time in hours |
| *Positive screening result to 4 months of age | 6 |
| From 4 months | 8 |
| From 8 months | 10 |
| From 12 months onwards | 12 |
* Around 2 weeks of age.
Source: www.bimdg.org.
Children with MCADD need to avoid medium chain triglycerides (MCT). Medium chain fatty acids can enter mitochondria without being bound to carnitine, so bypassing the step at which fatty acid oxidation is normally regulated (CPTI). MCT ingestion could therefore lead to a more severe metabolic disturbance than with long chain fats.
Dietary guidelines and information leaflets (e.g. MCADD is Suspected, MCADD and Your Child) for screened patients have been developed and can be accessed from www.bimdg.org.uk and www.newbornbloodspot.screening.nhs.uk. There are dietary information sheets for use at specified ages, from a positive screening result throughout childhood; they can also be used for clinically presenting patients. Information on suggested safe fasting times and ER for management of illness is included. Another useful educational tool is an e-learning resource on MCADD which is specifically designed for dietitians and midwives, accessed from www.geneticseducation.nhs.uk.
Dietary management during illness
If a child with MCADD is unwell (particularly if there is fever, vomiting and/or diarrhoea) and has a reduced appetite they should be given the standard ER of at least 3 hourly drinks containing glucose polymer, day and night (p. 502) . The ER needs to be implemented at an early stage to prevent metabolic decompensation and risk of sudden death. Fat is usually excluded during the acute period, although there is little evidence to support if this is essential. As the child improves, the normal diet can be resumed but extra ER drinks should be given, particularly during the night, until the child is fully recovered and eating well. If the ER is refused or vomited the child should have rapid access to their local paediatric services, avoiding a wait in the accident and emergency department, so there is no delay in starting an intravenous (IV) infusion containing glucose; 10% glucose, 0.45% saline is generally suitable but electrolytes should be monitored. Management decisions should be based on the child’s clinical state. Monitoring of blood glucose at home is not recommended because precision is poor at low concentrations and hypoglycaemia is a relatively late finding during illness in MCADD [16]. If for any reason the child needs to be commenced on a special infant formula or paediatric enteral feed during illness, it should not contain added MCT (see below). Emergency regimen guidelines (IV and oral) for hospital use can be accessed from www.bimdg.org.uk Although newborn screening has markedly improved the outcome for children with MCADD [17], there is still a risk of sudden death, specifically associated with vomiting [18]. The child who is vomiting should therefore be managed with extra vigilance.
Dietary management of well infants
With the introduction of newborn screening for MCADD in the UK it was important to set ‘maximum safe fasting times’ (Table 19.1). These were based mainly on feeding practices used in screened populations elsewhere in the world that had proven successful in avoiding symptomatic episodes. A review of these guidelines are planned
Infants can be breast or bottle fed with a normal infant formula and weaned onto a normal diet without restriction of long chain triglycerides (LCT), but it is important they are fed at regular intervals day and night. Small amounts of MCT are present in the vegetable oils of normal infant formulas, but the quantities are not sufficient to cause a problem. However, MCT are added to some special infant formulas and energy supplements and these should not be given. It is important to check the current composition of all such products for added MCT. Examples include hydrolysed infant formulas Pregestimil Lipil, MCT Pepdite, Pepti-Junior; high MCT special infant formulas Monogen, Lipistart; high MCT energy supplements MCT Duocal, MCT Procal. Low birthweight infant formulas should also be checked for added MCT.
The following dietary guidelines should be followed when treating an infant with MCADD:
Infants (under 1 year of age)
- breast feeding or normal infant formula is suitable
- avoidance of long fasts (Table 19.1)
- demand feeding every 3–4 hours throughout the day
- night feeds according to guidelines for ‘maximum safe fasting times’ (Table 19.1)
- weaning is commenced at the normal time around 6 months (26 weeks) of age
- weaning onto a normal diet and encouragement of regular starchy foods as weaning progresses
- restriction of LCT is not necessary
- avoidance of special infant formulas which contain added MCT
Note that newborn infants at risk for MCADD because of family history must receive adequate feeds (as described above).
Dietary management of well children
Children with MCADD should be encouraged to adopt a ‘healthy lifestyle’ through diet and exercise. Some parents worry their child may not be eating enough but they need to be reassured that not always finishing a meal can be normal; if necessary a snack can be offered later. There may be a risk of overweight or obesity due to parental anxiety surrounding feeding [19, 20]. Children with MCADD should be encouraged to exercise as normal. The need for provision of additional CHO pre-exercise should be individually monitored as muscle pain and/or reduced exercise tolerance has been reported in some patients [19]. All children, including those with MCADD, can feel hungry after exercise and a CHO snack should be given to help replenish glycogen stores.
From 1 year of age children can fast for 12 hours at night. In the author’s centre a minority of patients are not able to fast all night as they develop early morning symptoms; these symptoms have been resolved by being given either a night feed or uncooked cornstarch before bed as it is slowly digested to release glucose.
It is not necessary to restrict dietary fat. MCT, C8 and C10, occur in only a few foods and in small amounts, e.g. in butter, cow’s milk, and these can be included in the diet. Coconut is the only exception where 5% of the fatty acids have a chain length of C8 or C10; for coconut oil, the figure is 13%. Precautions should be taken in countries, such as the Philippines, where coconut oil is used as the main cooking oil. Pure coconut and coconut oil may be best avoided as the effect of large amounts of these in children with MCADD is not known. Theoretically there is risk of accumulation of toxic medium chain fats. Coconut products such as coconut milk and coconut paste may be suitable (provided the C8 and C10 content is low) and small amounts of coconut as an ingredient in food are acceptable in healthy children with MCADD. MCT are added to some special paediatric feeds and energy supplements and these should not be given. It is important to check the current composition of all such products for added MCT. Examples include hydrolysed paediatric feeds Peptamen Junior, Paediasure Pepdite; paediatric enteral feeds Paediasure, Frebini Energy; high MCT energy supplements MCT Duocal, MCT Procal.
The following dietary guidelines should be followed when treating a child with MCADD:
Children (from 1 year of age)
- regular meals (three main meals daily, including breakfast which must not be missed)
- a bedtime snack containing starchy foods such as bread, crumpets, muffins, rice cakes, crackers, biscuits, pitta bread, chapatti or cereals
- missed meals can be replaced with a starchy snack if necessary
- avoidance of special dietetic products which contain added MCT
- a maximum fasting interval of 12 hours overnight (Table 19.1)
- encouragement of a ‘healthy lifestyle’ and maintenance of normal body mass index
- restriction of LCT is not necessary.
Alcohol
Adolescents (and adults) need to know that an excessive alcohol intake is hazardous. Alcohol intoxication with resultant vomiting has been a cause of presentation in adults [8]. Vomiting and loss of appetite associated with excessive alcohol intake can be particularly dangerous. Alcohol inhibits gluconeogenesis so both fatty acid oxidation and gluconeogenesis which provide fuel during fasting are impaired. Moreover, the symptoms of hypoglycaemia might be attributed to inebriation rather than MCADD. Teenagers and adults need to be educated about the risks associated with excessive alcohol intake. A sensible approach is to advise that alcohol intake is limited and must always be taken in combination with food.
Carnitine
Plasma carnitine concentrations are often below the normal range in patients with MCADD, particularly after illness. The consequences of this are not clear. Carnitine supplements are given by some but this is not universal [21]. Maintenance of free carnitine levels (albeit at the lower end of normal range) has been reported in 46 patients from one centre not receiving carnitine supplementation; no patient experienced significant episodes of rhabdomyolysis or decompensation [22]. In the author’s centre, a trial of carnitine is given to a minority of children who complain of muscle symptoms and also have low plasma carnitine levels. There is conflicting evidence concerning the beneficial effect of carnitine on exercise tolerance [23, 24].
Disorders of long chain fatty acid oxidation
The long chain fatty acid oxidation disorders include:
- Defects of the carnitine cycle. Carnitine palmitoyl transferases I and II (CPTI and CPTII), carnitine acylcarnitine translocase (CACT) deficiencies and defects of the transporter for uptake of carnitine across the cell membrane (OCTN2 or CT) (Fig. 19.1).
- Defects of β-oxidation. Very long chain acyl-CoA dehydrogenase deficiency (VLCADD), mitochondrial trifunctional protein deficiency (MTPD) which encompasses the three MTP enzymes LCHAD deficiency (LCHADD), due to an isolated deficiency of LCHAD enzyme within the MTP complex (Fig. 19.1).
For a description of the biochemical pathway of long chain fatty acid oxidation refer to Introduction to Inherited Metabolic Disorders (p. 383), page 589 and Fig. 19.1.
Overview of clinical presentation
Long chain fatty acid oxidation disorders (LC-FAOD) vary in severity and may present at any time from the neonatal period to adulthood. The organs affected are heart, liver and skeletal muscles. The clinical presentation for all disorders commonly includes one or all of these problems:
- an acute ‘hypoketotic’ hypoglycaemia and encephalopathy with associated liver failure and hyperammonaemia
- cardiomyopathy and arrhythmias (often in infancy)
- myopathy presenting as weakness or episodes of muscle breakdown (rhabdomyolysis) induced by illness in early childhood or more commonly by exercise in older children or adults (as seen in patients with partial deficiency of CPTII); additionally some patients with CPTI deficiency have renal tubular acidosis during infancy
- most patients with LCHADD or MTPD develop pigmentary retinopathy and/or peripheral neuropathy as long term complications
- patients with partial deficiencies (e.g. VLCADD) often remain asymptomatic throughout childhood and sometimes throughout life
- most patients with LCHADD or MTPD develop pigmentary retinopathy and/or peripheral neuropathy as long term complications
A review of 187 patients with LC-FAOD identified hepatic symptoms (hepatomegaly, increased blood alanine transaminase (ALT) and steatosis), hyperammonaemia and hyperlactacidaemia as being the most predominant clinical and biochemical features at diagnosis, irrespective of the underlying defect [25]. More details of the clinical presentation of the individual disorders are described elsewhere [26]. Inadequate energy production from fatty acids and ketones and the accumulation of toxic intermediates (such as long chain acylcarnitines) are probably responsible for the clinical features [4]. Many of these symptoms are reversible with the provision of adequate, appropriate energy.
Genetics
All LC-FAOD are autosomal recessive inherited disorders. In all, there is molecular heterogeneity but prevalent mutations have been identified in some, e.g. CPTI (Inuit population only) and CPTII, LCHAD deficiencies. In different LC-FAOD the genotype and phenotype relationship varies, e.g. in VLCADD homozygous nonsense mutations are generally associated with severe early onset disease; the c.848T>C mutation has only been found in mildly affected or asymptomatic patients. A more detailed description of genetics for different LC-FAOD is found elsewhere [26, 27].
Expanded newborn screening
A number of countries worldwide such as Germany, Australia and the USA undertake expanded newborn screening for LC-FAOD, using tandem mass spectrometry. Expanded newborn screening for VLCADD has identified a common milder phenotype for which less dietary modification is required (see below). A pilot project of expanded newborn screening has been under way since July 2012 in parts of England; LCHADD is one of the disorders being evaluated. Screening is by measurement of hydroxypalmitoyl (C16-OH) carnitine in dried blood spots using tandem mass spectrometry. More information on the screening, diagnostic and clinical management protocols can be accessed at www.expandedscreening.org. LCHADD is not part of the national screening programme in England.
Principles of dietary management
Treatment of LC-FAOD disorders is primarily by diet with the exception of defects of the transporter for cellular carnitine uptake (OCTN2). Although they are different disorders the principles of dietary treatment are similar. The prescribed dietary treatment will vary depending on the severity, age at onset and underlying disorder. The main aims of dietary treatment are to prevent an energy deficit and, at the site of the defect, the accumulation of potentially toxic acyl-CoA intermediates and acylcarnitines (formed from these) by minimising fatty acid oxidation and provision of adequate energy. The abnormal metabolites which accumulate are disorder specific and present in higher concentrations during times of metabolic stress such as illness. Abnormal plasma acylcarnitines are not seen in CPTI or OCTN2 [26].
Long chain fatty acids for oxidation are derived from both dietary fat and adipose tissue. Dietary treatment therefore involves avoiding long fasts (especially during illness), exercise management (when energy needs are increased) and additionally in severe defects restricting the intake of dietary fat and instituting more frequent feeding with an increased CHO intake and supplements of MCT. The response to dietary treatment is mainly good. Cardiomyopathy (if present) will resolve and hypoglycaemia and major metabolic decompensations can be prevented. Muscle pain with exercise may still occur in some disorders, despite good dietary management. Patients with LCHADD and MTPD have specific long term complications, particularly a pigmentary retinopathy and progressive peripheral neuropathy (which is more of a problem in MTPD). It is reported that diet may help delay the onset and progression but not prevent retinopathy [28–30]. Frequent decompensations, late diagnosis with severe symptoms and hypoglycaemia may increase the progression of pigmentary retinopathy [29]. The pathogenesis of retinopathy remains to be fully elucidated.
During illness patients are at risk of metabolic decompensation due to increased lipolysis and fatty acid oxidation. Provision of glucose using a standard ER is needed to help prevent this.
Treatment recommendations have been published for β-oxidation defects based mainly on expert opinion from a European workshop [31, 32] and ‘A Delphi clinical practice protocol for the management of VLCADD’ from the USA [33]. Genetics Metabolic Dietitians International (GMDI) has also produced guidelines for management of VLCADD which can be accessed at www.gmdi.org. There are no such publications for defects of the carnitine cycle.
Long chain triglycerides
The safe upper limit for long chain fat intake is unknown and varies with the severity of the disorder. Normalisation of acylcarnitine profiles is observed in LCHADD and MTPD when LCT intake is <10% total energy and MCT are given [34]. Recommendations from published guidelines suggest for severe VLCADD to limit LCT to 5%–10% total daily energy intake [32, 33]. This is also recommended by GMDI. For severe defects of LCHADD and MTPD it is recommended to restrict LCT to as low as practically possible (5%–10% energy intake) but in practice it is typically more likely to be <15% (including 3%–4% from essential fatty acids) [32, 34]. Importantly, in those presenting with a milder myopathic form of the disorder, as can be seen in VLCADD and CPTII deficiency (CPTIID), such restriction of LCT may not be necessary [32].
Medium chain triglycerides
Dietary MCT are absorbed through the hepatic portal vein; they pass directly into the mitochondria primarily independent of the carnitine cycle and are then rapidly converted to ketone bodies, bypassing the long chain β-oxidation enzymes. Ketones inhibit the mobilisation of fatty acids from adipose tissue and the oxidation of fatty acids by cardiac muscle [35]. In patients with VLCADD, MTPD and LCHADD, MCT (C8, C10) can provide a useful energy source, 8.3 kcal (35 kJ)/g, and may have other beneficial effects. MCT have led to the resolution of cardiomyopathy in patients with VLCADD and LCHADD deficiencies [36, 37]. Medium chain fatty acids may also suppress long chain fatty acid oxidation without being converted to ketone bodies; they have been shown to inhibit the accumulation of potentially toxic long chain intermediates in cultured skin fibroblasts from LCHAD and MTP deficient patients [38]. It has been suggested that patients should maintain a high intake of MCT throughout life (10%–20% energy intake) [34]. The optimal amount or ratio of C8, C10 fatty acids to provide is not known. Skin fibroblast studies have shown that C10 undergoes elongation prior to oxidation, so it has been suggested that C8 may be preferable [39, 40]. Recommendations from published guidelines from GMDI and other authors [32, 33] suggest 20%–25% total daily energy intake be provided from MCT for severe LCHADD, MTPD and VLCADD. In the UK, infant formulas which can be prescribed for LC-FAOD provide either 17% or 30% total energy intake as MCT (Table 19.2).
Table 19.2 Low LCT infant formulas per 100 mL
| Monogen | Lipistart | Basic-f | Low Fat Module | ||
| (SHS) | (Vitaflo) | (Milupa) | (SHS) | ||
| Dilution | 17.5% | 15% | 13% | 18% | |
| Scoop size | 5 g | 5 g | 4.3 g | 6 g | |
| Energy | kcal | 73.5 (310 kJ) | 68 (282 kJ) | 49 (207 kJ) | 67.3 (286 kJ) |
| Protein | g | 2.2 | 2.1 | 1.8 | 1.6 |
| Carbohydrate | g | 12 | 8.3 | 10.2 | 14.9 |
| Fat | g | 1.9 | 3.1 | <0.1 | 0.14 |
| – MCT | g | 1.52 | 2.5 | Nil | Nil |
| % fat energy | 80% | 80% | |||
| % total energy | 17% | 30% | |||
| – C8 | g | 0.83 | 1.5 | ||
| – C10 | g | 0.6 | 0.93 | ||
| – LCT | g | 0.38 | 0.57 | ||
| % fat energy | 20% | 20% | |||
| – Linoleic acid | mg | 90 | 259 | Nil | Nil |
| % total energy | 1.1% | 3.4% | |||
| – α-linolenic acid | mg | 10 | 36.3 | Nil | Nil |
| % total energy | 0.17 | 0.5% | |||
| – DHA | mg | Nil | 30.3 | Nil | Nil |
| – AA | mg | Nil | 15.2 | Nil | Nil |
| Vitamins | |||||
| – E | mg | 0.5 | 1.7 | 0.3 | 0.4 |
| – A | µg | 56.9 | 69.9 | 52 | 71.3 |
| – D | µg | 1.2 | 1.4 | 0.9 | 1.2 |
Reconstitution: Monogen 1 scoop added to 25 mL water
Lipistart 1 scoop added to 30 mL water
In UK only Monogen and Lipistart are prescribable.
SHS, Scientific Hospital Supplies International; MCT, medium chain triglycerides; LCT, long chain triglycerides; DHA, docosahexaenoic acid; AA, arachidonic acid.
Source: Manufacturers.
The role of MCT is less clear in defects of the carnitine cycle. In several infants with CPTI deficiency, renal tubular acidosis has only resolved when they have been changed to an MCT based feed [41]. MCT has also been used with apparent benefit in patients with mild CACT deficiency (CACTD). In cases of severe CACTD and CPTIID, MCT has been reported to have caused markedly abnormal blood acylcarnitines and dicarboxylic aciduria [42, 43]. They suggest the amount of MCT, may need to be limited because it may in part enter the mitochondria by a carnitine dependent mechanism.
MCT is found in insignificant amounts in normal diets. Although MCT have benefits in LC-FAOD, their long term use in humans has never been systematically studied, so it is not known if there are any detrimental effects.
MCT and exercise
In the normal population the primary fuel source during low intensity prolonged exercise is fatty acids, and for high intensity exercise CHO (glucose and glycogen stores). Glucose is also the initial fuel during any exercise. Patients with VLCADD, LCHADD, MTPD and CPTIID can experience muscle pain and rhabdomyolysis with exercise. This may be due to their limited ability to oxidise long chain fatty acids as a fuel source for exercising muscle or the accumulation of toxic intermediates. Supplements of MCT pre-exercise have been reported to improve exercise capacity although only very small numbers of patients of varying ages and disorders have been studied [44–46]. In patients with the myopathic form of CPTIID and VLCADD there appears to be sufficient enzyme activity for fatty acid oxidation at rest, but not for the increased requirement of exercise [47, 48]. Some patients with CPTIID appear to benefit from taking a diet rich in polysaccharides pre-exercise [49]. As yet, no treatment has conclusively been shown to prevent rhabdomyolysis.
The effect of diet on exercise capacity has also been studied in the VLCADD knockout mouse. This has reduced exercise intolerance which is not improved by a low fat, CHO enriched diet [50]. Exercise capacity was improved by an MCT bolus prior to exercise but not by long term use of an MCT based diet. Moreover, long term use of MCT caused severe hepatic steatosis and increased long chain acylcarnitine accumulation [51] which was not seen following an MCT bolus [52]. Although these results are interesting it is not yet clear how they relate to human patients.
Triheptanoin
Triheptanoin, an oil containing C7 fatty acids, has been substituted for conventional MCT in a number of patients with LC-FAOD. Use of MCT leads to the production of acetyl-CoA but it is hypothesised that a shortage of oxaloacetate prevents this from being oxidised in the citric acid cycle. Triheptanoin generates acetyl-CoA and also propionyl-CoA. The latter can be converted to oxaloacetate, which acts as an ‘anaplerotic’ substrate for the citric acid cycle. Improved cardiac function and/or muscle weakness has been reported in three patients with VLCADD [53] and seven patients with myopathic CPTIID [54] given triheptanoin. However, triheptanoin was not compared with MCT in a controlled manner, so it is impossible to be certain if it conferred any specific benefit. Triheptanoin is not yet widely available outside the USA.
Frequency of feeding
Frequent feeding is recommended to reduce lipolysis. The safe duration of fasting for different disorders and ages has not been well defined and practices differ between countries and centres. Treatment recommendations from Spiekerkoetter et al. suggest a maximum safe fasting time at night of 8 hours from 6 months of age and 10 (−12) hours from 12 months in VLCADD, MTPD and LCHADD during stable metabolic condition [32]. In the USA no consensus on overnight feeding could be reached by an expert panel using the Delphi method [33]. The GMDI guidelines (www.gmdi.org) for VLCADD are similar to those of Speikerkoetter et al. In the UK, fasting is more severely limited to help reduce the risk of cardiomyopathy and onset of long term complications of retinopathy and neuropathy (although this has never been proved). Children with severe defects are fed 3–4 hourly during the day and by continuous nasogastric or gastrostomy feeding overnight (or the child is woken for feeds during the night). Overnight feeding is not standard practice but the literature suggests that some other countries do this, such as Scandinavia.
Questionnaire surveys have shown that uncooked cornstarch (UCCS) is used by some centres to provide a source of ‘slow release glucose’ [31, 55]. In older children continuous tube feeding overnight may be replaced with UCCS (personal practice). This may be a better option than stopping overnight feeds completely. Before use, the metabolic response to UCCS should be assessed since this varies between patients [56]. However, there are no published studies comparing the metabolic profiles of patients on overnight feeds, UCCS and those who fast overnight or the outcome between those given long term overnight feeding or not. Patients with milder defects are likely to tolerate overnight fasting without problems.
Prevention of deficiencies on a low LCT diet
Patients on minimal LCT diets require supplements of fat soluble vitamins, essential fatty acids (EFA) and long chain polyunsaturated fatty acids (LCPUFA). In the UK at least 1% total energy intake from linoleic acid and 0.2% from α-linolenic acid are recommended [57]. Others recommend a much higher intake of 4.5% daily energy intake in infants <4 months of age, decreasing to 3% from age 4 years [32]. The joint FAO/WHO 2008 report recommended an adequate daily intake for children >6 months to be linoleic acid 3%–4% and α-linolenic 0.4%–0.6% of daily energy intake [58]. Most sources of EFA (such as walnut oil) do not provide LCPUFA, such as arachidonic acid (AA) and docosahexaenoic acids (DHA), but these are expected to be synthesised from the EFA that are provided. In the author’s experience, deficiency is rare if patients are receiving adequate EFA in the form of walnut oil [59].
It has been suggested that DHA deficiency may contribute to the pathogenesis of chorioretinopathy of LCHADD [60], although this is not prevented by supplementation. Nevertheless supplements of DHA at a dose of 60 mg/day in children <20 kg and 120 mg/day in children >20 kg are recommended in MTPD and LCHADD [32]. FAO/WHO recommends an adequate daily intake of DHA for normal children to be
- the amounts found in breast milk for infants
- 100–150 mg for children aged 2–4 years
- 150–200 mg for children aged 4–6 years
- 200–300 mg for children aged 6–10 years [58]
Fat soluble vitamin supplements (A, D, E) are essential for those on reduced LCT intake. Vitamin E deficiency is associated with a peripheral neuropathy which is one of the long term complications of MTPD and LCHADD. It is therefore essential that vitamin E is provided in normal requirements for age, at least.
Carnitine
It remains uncertain whether carnitine supplements should be given to patients with long chain fatty acid oxidation disorders. Plasma free carnitine concentrations are often below the normal range, particularly after episodes of illness, but tissue levels are probably not low enough to affect fatty acid oxidation. Supplements may facilitate the excretion of metabolites but, by increasing the level of long chain acylcarnitines, it is possible that they may increase the risk of arrhythmias [61].
Energy distribution of the diet
As LCT is restricted more energy needs to be provided from CHO and supplements of MCT. The optimal distribution of energy intake from CHO and MCT is not known. The following provides a guide to the typical energy distribution and long chain fat intake of the diet in severe defects (based on personal practice):
- 60%–65% energy from CHO
- 10%–15% energy from protein
- 20%–25% energy from medium chain fat (depending on the disorder)
- 1.2%–2% energy from EFA (based on FAO/WHO [58] higher intakes may be appropriate)
Long chain fat
During infancy, total LCT intake will vary depending on the choice of low LCT infant formula as the percentage energy from LCT varies (Table 19.2): Monogen 4.5%, 0.5 g LCT/100 kcal (420 kJ); Lipistart 6.9%, 0.8 g LCT/100 kcal (420 kJ). However, a source of LCPUFA needs to be given with Monogen, so the overall LCT intake will increase. The LCT intake from formula could range from 2 to 7 g/day in infants aged <6 months. In children, it is possible to restrict intake of long chain fat to provide ≤ 6% total energy intake, ≤7 g LCT/1000 kcal (4.2 MJ), excluding EFA. Similar dietary practices are reported to be used by others [28, 32].
The suggested percentage of energy from protein is based on typical intakes in the normal UK population. Due to concerns about excess weight gain over time in their cohort of LCHADD and MTPD patients, Gillingham et al. trialled a high protein diet providing 30% energy intake in an attempt to lower energy intake, without loss of metabolic control [62]. Short term, the diet was reported to achieve this and increase resting energy expenditure. Longer term studies are needed to see if this can be sustained.
Minimal long chain fat diet
Infants
Infants with severe long chain defects need a minimal LCT feed supplemented with MCT and frequent feeding. This could be provided by Monogen or Lipistart; both are nutritionally complete infant formulas with minimal LCT (Table 19.2). Exclusive breast feeding is contraindicated except for asymptomatic VLCADD infants diagnosed on newborn screening. Compared with Monogen, energy from MCT is higher in Lipistart and energy from CHO is lower; it also contains LCPUFA and provides more vitamin E. It is important to establish if MCT can be oxidised before using either of these formulas. If MCT is considered inadvisable because of a possible diagnosis (as in, perhaps, severe CACTD or CPTIID), a modular feed without fat (long or medium chain) may need to be used during the initial stages of management until the diagnosis is established. Skimmed milk powder or whey protein powder, although they do contain small amounts of LCT, can be used as the protein base with glucose polymer and additional minerals and vitamins to provide recommended intakes. EFA and LCPUFA should also be provided if the infant remains on a modular feed for several days. Suitable commercial minimal fat feeds with no added MCT are Low Fat Module (0.14 g fat/100 mL) or Basic-f (<0.1 g fat/100 mL), although the latter is generally not available in the UK (Table 19.2).
Clinically diagnosed infants are likely to be very sick, managed in intensive care and receiving IV 10% dextrose. Once stabilised, nasogastric (or oral) feeds should be introduced and titrated against IV fluids so that an adequate energy intake is maintained and further metabolic decompensation is prevented. The initial feeds given may vary depending on the clinical situation but should, if possible, provide 10 g CHO/100 mL (10% CHO) to maximise energy intake and reduce lipolysis. Once the infant is well and established on full feeds these should be given 3 hourly during the day and night, or 3 hourly during the day with a continuous feed overnight. The overnight feed can be commenced 2 hours after the last 3 hourly day feed and can probably be stopped 2 hours before the first day feed in the morning. The amount of glucose required overnight to minimise lipolysis is not known. For infants, practice is to provide a minimum of 0.5 g CHO/kg/hour which will equal basal glucose production rates (p. 544) and should therefore minimise lipolysis. When babies are just on infant formula the feed is distributed evenly over the 24 hours, e.g. a 6 kg infant fed 150 mL/kg (900 mL) would have 120 mL 3 hourly × 5 feeds during the day (at 8 am, 11 am, 2 pm, 5 pm, 8 pm) with 300 mL given at 40 mL/hour continuously overnight for 8 hours from 10 pm to 6 am. At this volume Monogen provides 0.8 g CHO/kg/hour and Lipistart 0.5 g CHO/kg/hour (but provides more energy from MCT than Monogen).
Infants diagnosed on newborn screening
Treatment recommendations have been given by Spiekerkoetter et al. for infants diagnosed on expanded newborn screening [32, 61]. The dietary treatment advised for VLCADD depends on the clinical presentation and biochemistry. Asymptomatic infants with a normal creatine kinase and liver transaminase levels can be fed normally, either breast fed or with standard infant formula. In symptomatic infants, or those with abnormal creatine kinase or transaminases, the diet should contain greater amounts of MCT formula and less or no breast milk. If cardiomyopathy is present then feeding should proceed as for the clinically presenting patient. Asymptomatic infants should continue to have clinical and biochemical follow-up.
For LCHADD (MTPD) dietary treatment should proceed as for the clinically presenting patient, even if the infant is asymptomatic. There is some evidence that this may delay the onset and progression of the long term complications of peripheral neuropathy and retinopathy.
Weaning
Weaning is commenced at the normal time around 6 months (26 weeks) of age. An example of a suggested menu plan for infants is provided in Table 19.3. Solids should have a minimal fat content and be high in CHO. Rice cereal, potato, fruit and vegetables are suitable as first weaning foods. Once these have been introduced low fat, high protein foods can also be given, e.g. turkey, white fish, lentils, very low fat cottage cheese and yoghurt. Commercial baby foods can be included in the diet but need to be limited to those with a low fat content. Wet baby foods (fat content <0.5 g/100 g) and dried baby foods (fat content <2 g/100 g) can be given freely. Baby foods with a higher fat content need to be counted as part of the daily fat intake. Parents can be taught how to calculate the amount of fat a food provides from the nutritional labelling (p. 602) . As the amount of starchy solid food begins to increase, this should replace some of the 3 hourly day feeds, ensuring it provides at least the same amount of energy, e.g. an 8 kg infant fed 150 mL 3 hourly × 5 daytime feeds (8 am, 11 am, 2 pm, 5 pm, 8 pm) and 50 mL/hour continuous overnight feeds from 10 pm to 6 am. Each 150 mL of minimal fat feed provides 110 kcal (460 kJ) so the replacement solids should provide the same energy. Alternatively this can be calculated as follows:
- energy requirements are 95 kcal (400 kJ)/kg = 760 kcal (3.2 MJ)
- provide 60% of daily energy requirement from CHO = 460 kcal (1.9 MJ) = 5 g CHO/hour = 15 g CHO for 3 hours (0.6 g CHO/kg/hour)
Table 19.3 Minimal long chain fat weaning diet: sample menu
| 8 am Breakfast | *Commercial baby cereal mixed with Lipistart or Monogen |
| 11 am | Infant feed |
| 1 pm Lunch | Purée turkey, chicken (white only), white fish or lentils |
| Purée or mashed potato, sweet potato | |
| Pasta, rice | |
| Purée or mashed vegetables | |
| *Commercial baby savoury foods | |
| 4 pm Tea | Purée or mashed fruit and sugar or *commercial baby fruit dessert |
| Very low fat yoghurt or fromage frais | |
| Milk pudding (custard, cornflour, ground rice) made with infant feed or skimmed milk | |
| 7 pm | Infant feed |
| 9 pm to 6 am | Continuous overnight feed for a 7 kg infant: |
| Monogen – 30 mL/hour provides 0.5 g CHO/kg/hour (197 kcal, 825 kJ) | |
| Lipistart – 40 mL/hour provides 0.5 g CHO/kg/hour (245 kcal, 1025 kJ, provides) | |
| Lipistart provides more MCT so a lower volume could be considered | |
| Extra feeds or baby juice can be given with meals | |
| Finger foods may be introduced from 6–9 months: soft cooked vegetables, soft ripe fruit (pasta, bread and cereals may also be suitable but LCT content must be checked first) | |
* Commercial baby foods.
Allow freely: wet baby foods ≤0.5 g fat/100 g and dried baby foods ≤2 g/100 g.
Wet baby foods 0.5–1.0 g/100 g or dried baby foods 2–5 g/100 g may also be suitable but counted as part of total fat intake.
Baby foods with a higher fat content are best avoided.
Walnut oil dose (p. 601) .
Infants who eat well will easily exceed this amount of CHO in a meal but the amount of solids taken needs to be checked in those who feed poorly. MCT oil (p. 602) can be incorporated into the diet of older infants who are eating well. The infant formula can continue to be given overnight, but with increasing age larger volumes are needed to provide the required amount of CHO; this may interfere with the daytime appetite or lead to excessive weight gain. Therefore as the amount of solids progresses the volume of feed should be decreased and glucose polymer added to provide the required amount of CHO (0.5 g CHO/kg/hour = 2 kcal (8 kJ)/kg/hour). It is not yet clear whether the overnight feed should be changed to glucose polymer or whether it is helpful to continue some MCT at night [63]. In this case it may be more appropriate to base the overnight feed on a total energy intake per kilogram per hour from CHO and MCT, although this is not current practice.
Fat soluble vitamin supplements will be required once the daily intake of certain infant formula falls below 600 mL (Table . p. 602).
EFA intake differs depending on the formula used (Table 19.2). Once weaning is commenced and volume of formula decreases and no longer provides the sole source of nutrition, an additional source of EFA is needed. Walnut oil provides a good source of EFA and allows a minimum amount of LCT to be given (Table 19.4). To provide the UK suggested requirement of EFA the dose of walnut oil needed is 0.1 mL per 56 kcal (235 kJ) of the estimated average requirement (EAR) of energy for age [57], subtracting the energy provided by the feeds. If the infant is on Lipistart >1% of energy intake from linoleic acid will be given. The walnut oil is administered as a single dose and given as a medicine from a spoon or via the feeding tube. In the UK, walnut oil can be purchased from most large supermarkets. It should be stored as recommended to avoid peroxidation. Other oils such as sunflower, safflower (mainly linoleic acid), soya and flax are also rich sources of EFA and a combination of these can be used to give the required EFA intake if walnut oil is disliked.
Table 19.4 Walnut oil – essential fatty acid composition
| 1 mL walnut oil provides | |
| Energy | 8.4 kcal |
| 35 kJ | |
| Fat | 0.93 g |
| Linoleic acid | 0.58 g |
| α-linolenic acid | 0.12 g |
| Ratio n6:n3 fatty acids | 4.5 |
For analysis of other oils see Fatty acids, 7th Supplement to McCance and Widdowson’s The Composition of Foods, 5th edn. Cambridge: Royal Society of Chemistry and London: Ministry of Agriculture, Fisheries and Food, 1992 and p. 754.
LCPUFA are added to Lipistart; 400 mL provides the suggested intake of 60 mg/day advised for LCHADD [32]. Monogen does not contain added DHA or AA, so additional supplements are necessary. These can be added as KeyOmega (p. 630): one sachet provides 100 mg DHA and 200 mg AA.
Children
Long chain triglycerides
Children should remain on a minimal LCT diet with an energy distribution as previously outlined (p. 599) . Inevitably, daily intake of long chain fat increases with age. The figures quoted are based on the lowest possible long chain fat intake that is practicably achievable for a given age, as the safe upper limit for long chain fat intake is not known. Ideally, the increased CHO intake should be derived from starchy foods (e.g. rice, pasta, potato, bread, cereals) because these are more slowly digested than sugary foods. It is recognised, however, that some children cannot manage such a bulky diet and sugar containing foods such as low fat ice cream, cake or biscuits should form part of their diet. The diet should contain very low fat sources of protein, such as white fish, white chicken or turkey meat, pulses (Table 19.5). There are many very low fat alternatives to regular high fat foods such as crisps, sauces, desserts, ice cream and cheeses which can also be incorporated into the diet. To allow a greater variety of manufactured foods to be included in the diet, parents need to be given guidance on interpreting food labels for fat content and on understanding the many and potentially misleading wordings used to describe the fat content of food, e.g. reduced fat, low fat, virtually fat free, 90% fat free. The different types of fat, e.g. saturated, mono-unsaturated and polyunsaturated, can cause confusion and it is helpful to explain that total fat should be used when calculating fat intake. Nutritional labelling expresses the total fat content as grams of fat per 100 g and often grams of fat per portion. To help parents work out if a food is suitable they can be taught to calculate and then weigh the amount of food which provides a certain amount of fat. This also makes it easier to monitor the daily fat intake. For example,

Table 19.5 Minimal long chain fat diet
| Foods allowed | Foods to avoid | |
| Milk | Skimmed milk, condensed skimmed milk | Full fat and semi-skimmed milk. Cream (all types) |
| Natural yoghurt, very low fat yoghurt and fromage frais (<0.2 g/100 g) | Full fat yoghurt and fromage frais | |
| Low fat cottage cheese (<0.5 g/100 g) | Full and half fat cheeses | |
| *Very low fat cheeses | ||
| Quark (skimmed milk soft cheese) | ||
| Low fat ice cream | †Ice cream | |
| Egg | Egg whites, egg white replacer, meringue | Egg yolks |
| Fish | White fish (no skin), e.g. haddock, cod, sole, plaice | Oily fish, e.g. sardines, kippers, salmon, mackerel |
| Crab, crabsticks, tuna, prawns, shrimps, lobster | Fish in breadcrumbs, batter, sauces, pastry | |
| Tinned: tuna in brine or water, crab, prawns, shrimps | Tinned: fish in oil | |
| Poultry | *Chicken, *turkey (white breast meat, no skin) | Chicken, turkey (dark meat and skin), basted poultry, duck, goose |
| Chicken in breadcrumbs, batter, sauces, pastry | ||
| Meat | *Lean red meat (<5% fat content) | Fatty meat, sausages (normal and low fat), burgers, meat paste, paté, salami, pies |
| Meat substitutes | Soya mince, *Quorn, *tofu | |
| Pulses | Peas, e.g. chick peas, split peas | None, unless in made-up dishes containing fat |
| Beans, e.g. red, white, borlotti, black-eyed | ||
| Fats/oils | Medium chain triglyceride oil as advised | Butter, margarine, low fat spread, vegetable oils, lard, dripping, suet, shortening |
| Pasta and rice | Spaghetti, macaroni, other pasta, noodles, couscous, rice (white) | Wholemeal pasta, pasta in dishes, e.g. macaroni cheese, carbonara |
| Brown rice, egg noodles | ||
| Flours and cereals | Flour (white), cornflour, custard powder, semolina, sago, tapioca | Flour (wholemeal), soya flour, oats, bran Foods made with flour which contain fat, e.g. pastry, sauces, cake, biscuits, batter, breadcrumb coatings |
| Breakfast cereals | Many are suitable | Cereals with nuts and chocolate, e.g. muesli |
| Wholewheat cereals, e.g. Weetabix | ||
| branflakes are higher in fat content than non-wholewheat cereals , e.g. Rice Krispies, | All-bran, Ready Brek | |
| cornflakes | ||
| Bread and crackers | *White bread, white pitta, crumpets, muffins Some crackers have a low fat content, e.g. rice cakes, Matzos, Ryvita (not sesame) | Wholemeal, wholegrain breads, nan bread, chapatti made with fat, croissants, oatcakes, cheese crackers, †crackers |
| Cakes and biscuits and pastry | Only those made from low fat ingredients 95% fat free cakes and biscuits | †Cakes, †biscuits, buns, pastry for sweet and savoury foods, e.g. apple pie, quiche |
| Desserts | Jelly, meringue, sorbet, very low fat ice cream, skimmed milk puddings, e.g. rice, custard | Most desserts, e.g. whole milk puddings, trifle, cheesecakes, gateaux, †mousse, fruit pie or crumble |
| Fruit | Most varieties – fresh, frozen, tinned, dried | Avocado pears, olives, yoghurt/chocolate covered dried fruit, banana chips |
| Vegetables | All vegetables and salad Very low fat crisps | Chips, †crisps, †low fat crisps, roast potato, potato or vegetable salad in mayonnaise or salad dressing, †coleslaw |
| Herbs and spices | Pickles, chutney Herbs, spices, salt, pepper | |
| Nuts Seeds | Nuts, peanut butter, seeds, e.g. sesame, sunflower | |
| Sauces and gravies | Tomato ketchup, brown sauce, soy sauce, Marmite, Oxo, Bovril, very low fat gravy mixes Fat free dressings and mayonnaise Minimal fat sauces (jars, tins, packets) | †Gravy granules, †stock cubes Salad cream, mayonnaise, oil and vinegar dressings †Sauce mixes (jars, tins, packets) |
| Soups | Some low calorie and ‘healthy eating type’ soups are very low fat | Most soups, cream soups |
| Confectionery | Boiled sweets, jelly sweets, fruit gums, pastilles, marshmallow, mints, ice lollies | Chocolate, chocolate covered sweets, toffee, fudge, butter mints |
| Sugars and preserves | Sugar, golden syrup, jam, marmalade, honey, treacle | Lemon curd, chocolate spread |
| Baking products | Baking powder, bicarbonate of soda, yeast, arrowroot, essences, food colouring | |
| Drinks | Fruit juice, squash, fizzy drinks, milkshake flavourings, tea, coffee | †Instant chocolate drinks, cocoa, malted milk type drinks, e.g. Horlicks |
* Intake of these foods may need to be restricted because of their fat content.
† These foods in the ‘avoid list’ often have very low fat equivalents which can be included in the diet.
Manufactured foods which have a fat content <0.5 g/100 g can generally be allowed in the diet freely, unless large amounts are being consumed.
Medium chain triglycerides
The intake of MCT naturally falls as less infant formula is consumed. Alternative sources of MCT should be incorporated into the diet because of the potential beneficial effects discussed above. A daily intake of 20%–25% of total energy intake is suggested which equates to 2.5–3 g/100 kcal (420 kJ) consumed. The optimal timing of administration has not been defined; however, it is common practice to divide the dose and give 3–4 times a day, as it is rapidly absorbed and used.
MCT can be added to the diet as an oil, emulsion or powder. These products vary in C8, C10 fatty acid composition (Table 19.6). MCT oil can increase the palatability of the diet. It has a low smoke point compared with other cooking oils. Care must be taken when cooking foods in MCT oil to ensure it does not burn or become overheated as it develops a bitter taste and an unpleasant odour. The optimum cooking temperature for MCT is 160°C. MCT oil can also be used in baking, e.g. cakes, biscuits and pastry, and can be added to a variety of foods such as pasta, soups. MCT emulsion such as Liquigen may be more acceptable as it can be easily mixed into skimmed milk without separating. It can also be added to other liquids or foods and used in baking. MCT Procal powder can be added to hot or cold food or drinks (not fruit juice as it curdles). MCT Procal comes in a convenient sachet form, making it easy to use away from home (p. 632). Recipes are available for all products from their manufacturers.
Table 19.6 Medium chain triglyceride products
| MCT g/100 g fatty acids | ||||||
| Product | Energy kcal (kJ) | MCT | C8 | C10 | C12 | Other nutrients |
| MCT oil (Nutricia) | 855 (3.6 MJ)/100 mL | 94 g/100 mL | 58 | 38 | 1.7 | Nil |
| Liquigen emulsion (Nutricia) | 450 (1.9 MJ)/100 mL | 50 g/100 mL | 82.1 | 15.9 | 1.3 | Nil |
| MCT Procal (Vitaflo) | 657 (2.7 MJ)/100 g 105 (440 kJ)/16 g sachet | 62.2 g/100 g 10 g/16 g sachet | 54 | 39 | – | Per sachet: 2 g protein, 3.3 g CHO |
MCT, medium chain triglyceride.
Other MCT products such as MCT margarine can be sourced from www.ceres-mct.com or www.drschar.com. These are not prescribable in the UK.
Table 19.7 gives an example of how to provide the daily intake of MCT.
Table 19.7 Example providing daily medium chain triglyceride intake
| Daily MCT intake for a 4-year-old girl | |
| Estimated average requirement for energy for age = 1400 kcal (5.9 MJ)/day [57] | |
| Provide 20%–25% energy from MCT = 280–350 kcal (1.2–1.5 MJ) ÷ (8.3 kcal, 35 kJ/g MCT) = 34–42 g/day | |
| Breakfast | 20 mL MCT emulsion (10 g MCT) added to breakfast cereal |
| Lunch | 1 sachet MCT Procal (10 g MCT) added to very low fat yoghurt at school |
| Evening meal | 10 mL MCT oil (10 g MCT) added to cooked pasta |
| Another 10 g of MCT could be added to the night feed (Table 19.8) from MCT emulsion | |
MCT, medium chain triglyceride.
Other nutrients
EFA need to be provided in the diet and can be added from walnut oil or combinations of other oils as described for infants. LCPUFA can be given as one sachet of KeyOmega, 100 mg DHA and 200 mg AA, which is more than the suggested intake of 60 mg DHA/day for young children with LCHADD or TFPD. In children weighing >20 kg more DHA is needed to provide the suggested intake of 120 mg/day [32]. One sachet of DocOmega provides 200 mg DHA.
Supplements of fat soluble vitamins (A, D and E) are needed unless the child is still taking around 600 mL infant formula. Intakes of iron, zinc and B12 should be regularly calculated as these can also be low in minimal LCT diets. The choice of supplement will depend on need, age and desired format: powder, tablets or soluble tablets are available. Ketovite tablets and liquid provide an adequate intake of fat soluble vitamins and are suitable for all ages. Alternatively, more complete vitamin and mineral supplementation can be provided: Fruitivits (from age 3 years), Paediatric Seravit (all ages), Phlexyvit sachets or tablets (from age 10 years), Forceval Soluble Junior (from age 6 years) and Forceval Soluble (from age 12 years) (p. 21) .
Frequency of feeding
Lipolysis can be minimised by 3–4 hourly feeding during the day and continuous overnight tube feeds. It is difficult to be sure, however, whether overnight feeding is necessary, particularly in older children who have larger glycogen stores. In LCHADD it may help delay the onset of retinopathy or neuropathy. If overnight feeding is used the amount of glucose given overnight should equal basal glucose production rates for age (around 0.3–0.4 g CHO/kg/hour) (p. 544) . It is not yet known whether the addition of MCT to the night feed is helpful [63]. Less CHO energy may be needed if MCT is added to the night feed. Table 19.8 gives examples of overnight feeds.
Table 19.8 Examples of an overnight feed for long chain fatty acid oxidation disorders
| Overnight feed for a 6-year-old boy, weight 22 kg |
| 25% CHO from glucose polymer, administered at 25 mL/hour from 10 pm to 6 am via gastrostomy |
| Fluid 200 mL |
| Energy 200 kcal (840 kJ) |
| Glucose polymer 53 g (95% CHO) |
| CHO 0.3 g CHO/kg/hour |
| or |
| Glucose polymer and MCT feed, administered at 25 mL/hour from 10 pm to 6 am via gastrostomy |
| Fluid 200 mL |
| Energy 200 kcal (840 kJ) |
| Glucose polymer 30 g (95% CHO) = 114 kcal, 479 kJ |
| MCT = ¼ of daily dose (10 g MCT) as 20 mL Liquigen (=5 g MCT/100 mL) = 90 kcal, 370 kJ |
MCT, medium chain triglyceride.
Fasting tolerance increases with age; therefore in older children it may be possible to stop the overnight feeds completely or substitute with one or more doses of UCCS. Before changing management, it is the author’s and others’ practice [56] to assess the child’s metabolic response by serial measurement of plasma free fatty acids, blood glucose and long chain 3-hydroxyacylcarnitines on their usual regimen and after a dose of UCCS. This needs to be done in hospital under controlled conditions, ensuring the child is not fasted for longer than planned. If UCCS is to be used the results of these studies are used to plan how frequently a dose of UCCS needs to be given. Doses of cornflour used are 1–1.5 g/kg/dose in older children. UCCS is generally introduced at home to test its palatability and tolerance. It should be given raw because cooking or heating disrupts the starch granules and thus makes it much less effective. It is usually mixed with skimmed milk. The dose of UCCS is gradually increased, starting with 5 g and increasing in 5 g increments every few days/week to the full amount.
Exercise management
Patients with VLCADD, LCHADD, MTPD and CPTIID can experience episodes of rhabdomyolysis, with myalgia, elevated creatine kinase levels and, in severe cases, myoglobinuria. These episodes are sometimes triggered by infection but more often they are induced by exercise in children or young adults as they become more active and play sports for longer periods. Rhabdomyolysis can be the only clinical symptom in patients with partial deficiencies of CPTII or VLCAD and management can be difficult. Under normal circumstances CHO is the main fuel used for moderate to high intensity exercise such as sprinting, whereas fatty acids are the main fuel for low to moderate intensity exercise. Thus ‘short burst exercise’ may be more suitable for these patients than prolonged low intensity exercise because CHO will be used in preference to long chain fatty acids. Several papers suggest that dietary modification may reduce the risk of rhabdomyolysis. In one study patients with partial CPTIID tolerated exercise better on a CHO rich diet than on a fat rich diet [49]; a bolus of glucose before exercise did not help [64]. In contrast pre-exercise boluses of MCT (0.3–0.4 g/kg/dose) have been reported to improve exercise capacity in patients with various fatty acid oxidation disorders [65]. Pre-exercise dietary treatment may need to be adjusted according to the intensity and duration of the exercise being undertaken. MCT can be given as a prescribed dose of Liquigen, or MCT Procal, and CHO can be given from food such as a low fat cereal bar, banana, bread or high CHO drink such as Lucozade. Some children may not be able to do prolonged periods of exercise without developing muscle pain, so this must be avoided. In some patients, drug treatment with bezafibrate may also be helpful [66].
If a child develops rhabdomyolysis and muscle pain during or following exercise they may find it very difficult to walk for 24–48 hours. These episodes must be treated promptly as there is a risk of acute renal failure. An emergency regimen using the standard glucose polymer ER (p. 502) and extra fluids (determined on an individual basis), is implemented and may help reduce the risk of worsening rhabdomyolysis. MCT can be added to the ER (personal practice with a few patients). The ER feed should aim to provide at least the normal energy intake for age. The feed can be given orally but if there is associated loss of appetite then it should be given via a tube to ensure the full amount of fluid and energy is taken. Rest is important until symptoms resolve. It is essential parents and patients know to look for signs of myoglobinuria (dark, cola coloured urine) and make immediate contact with their metabolic team as a hospital admission may be necessary. Plasma creatine kinase can be markedly elevated during episodes of rhabdomyolysis and may be helpful in determining the extent of muscle damage and management needed.
Feeding problems
Feeding problems are common in children with LCHADD, particularly in early childhood, and they occur despite good metabolic and clinical condition [67]. With age these problems either improved or resolved in this cohort. However, early recognition and management of feeding problems remains important and newborn screening may help prevent the development of these.
Monitoring
Dietary monitoring
Once an infant or child is established on dietary treatment, families need continued support and advice. Regular monitoring of this complex diet is essential to
- ensure the diet is nutritionally adequate, in particular intakes of fat soluble vitamins, EFA and LCPUFA
- ensure the overnight feeds provide adequate CHO (or energy) for age
- check the intake of long and medium chain fat
- provide new ideas and information on low fat manufactured foods
- assess exercise tolerance and provide advice on CHO/MCT intake
- check the ER for CHO concentration and volume and parents’ understanding of its use
Clinical monitoring
This should include a review of weight, growth and development. There has been little documented on the growth of children with LC-FAOD. Gillingham et al. reported excess weight gain with time in most of their LCHADD and MTPD patients [62] but others have not observed this, including the author’s centre [56]. If a child is overweight this needs to be managed carefully as weight loss could precipitate metabolic decompensation. A gradual reduction in energy intake is needed. If a child develops muscle pain with exercise this can make weight management difficult. Cardiomyopathy usually resolves on institution of a minimal LCT diet supplemented with MCT. Thereafter cardiac function (echo and electrocardiogram) should be monitored annually. Liver ultrasound is also performed annually to assess for steatosis. In LCHADD and MTPD annual ophthalmological (electrophysiology) and neurophysiological follow-up is important. For LCHADD an ocular examination is suggested within the first month of diagnosis as changes have been documented as early as 4 months of age [30].
Biochemical monitoring
Blood acylcarnitine concentrations may correlate with outcome in these disorders, but this is not certain. For example in LCHAD deficient patients cumulative long chain 3-hydroxyacylcarnitine concentrations appear to correlate with the progression of retinal disease [30]. Monitoring of acylcarnitines may therefore be useful in guiding dietary management. Plasma free carnitine levels should also be monitored, although there is dispute about supplementing carnitine if levels are low (p. 598) , except in OCTN2. Plasma transaminases and creatine kinase are helpful markers of clinical status. Creatine kinase increases markedly during episodes of exercise induced rhabdomyolysis and returns to normal once treated. Fat soluble vitamins, EFA and LCPUFA concentrations should be monitored annually and more often if there are concerns about dietary deficiencies. Erythrocyte measurements are a better marker of long term nutritional status than plasma. Other nutritional markers such as iron and B12 status may also be necessary if there are concerns about nutritional adequacy of the diet.
Emergency regimen for treatment of illness in LC-FAOD
If the child becomes unwell and is unable to take their usual regimen the standard ER of very frequent feeds of glucose polymer, day and night, should be given (p. 502) . In mild illnesses it will probably not be necessary to implement the ER. In patients with a gastrostomy or nasogastric tube, the drinks can be given via the tube if refused orally. The ER needs to be started promptly to inhibit the mobilisation of fatty acids as decompensation may be rapid. Long chain fat is strongly contraindicated. The role of MCT during acute illness is not clear. As the child improves, the normal diet can be resumed. While this is being reintroduced it is essential to maximise energy intake and continue with frequent feeding, usually 2–3 hourly by day and continuous tube feeds overnight. If the child does not tolerate the ER enterally they need to be admitted to hospital for an IV infusion containing glucose; 10% glucose, 0.45% saline is generally suitable but electrolytes should be monitored. Emergency regimen guidelines (IV and oral) for hospital use can be accessed from www.bimdg.org.uk. IV carnitine should not be given because long chain acylcarnitines may be arrythmogenic.
Patients with cardiomyopathy should probably be admitted during any intercurrent illness since there is a risk of deteriorating cardiac function. Families should also be warned to look for dark urine since illness (or exercise) can precipitate rhabdomyolysis with myoglobinuria. If there is myoglobinuria it is essential that the child is admitted to hospital for further management because of the risk of acute renal failure. A high fluid intake is recommended to increase myoglobin excretion and help prevent renal failure. Blood glucose monitoring by parents is not recommended as a marker of metabolic status, since hypoglycaemia is a relatively late finding and treatment should be initiated long before this develops [16].
Carnitine transporter deficiency
Patients with carnitine transporter deficiency (CT or OCTN2, Fig 19.1) have low plasma and intracellular concentrations of carnitine, which can impair fatty acid oxidation as carnitine is necessary for the transport of long chain fatty acids into the mitochondria. Patients generally present between 1 and 7 years of age with the typical features of an LC-FAOD; other patients remain asymptomatic [26]. Heart failure is a common presentation which can rapidly deteriorate with risk of death if patients are not treated with carnitine (around 100 mg/kg/day divided into two or three doses). Doses are adjusted according to plasma carnitine level. When given carnitine, generally no dietary treatment is necessary except during illness when there is a risk of metabolic decompensation. The standard ER of frequent glucose polymer drinks day and night should be given to prevent increased lipolysis (p. 502) . A more detailed overview of clinical manifestations, diagnosis and management is described elsewhere [26, 68].
Electron transfer defects
Multiple acyl-CoA dehydrogenase deficiency (MADD), also known as glutaric aciduria type II is caused by defects of electron transfer flavoprotein (ETF) or ETF ubiquinone oxidoreductase (ETFQO, fig 19.1). These molecules pass electrons from a number of flavin adenine dinucleotide (FAD) containing dehydrogenase enzymes to coenzyme Q in the mitochondrial respiratory chain. The dehydrogenases include the acyl-CoA dehydrogenases of β-oxidation and also enzymes involved in choline metabolism and the breakdown of several amino acids (lysine, tryptophan and the branch chain amino acids).
MADD has a wide range of clinical severity. All forms of the disorder can cause hypoglycaemic encephalopathy. The most severely affected patients present with congenital malformations, such as renal cystic dysplasia leading to pulmonary hypoplasia (Potter’s syndrome). Less severely affected patients may present with cardiomyopathy (usually as neonates) or with muscle weakness (at any age). Sudden death can occur in these patients. Some mild cases show a clinical and biochemical response to riboflavin (100–200 mg/day), the co-factor for these enzymes and the acyl-CoA dehydrogenases [69]. These patients usually present with progressive muscle weakness as young adults although other symptoms such as cyclical vomiting may have occurred in childhood. Riboflavin responsiveness is usually associated with mutations affecting ETFQO. Patients with late onset ETFQO deficiency have also been shown to have a secondary coenzyme Q10 (CoQ10) deficiency and supplements are recommended [70]. A more detailed description of the biochemical and clinical presentation of the individual disorders is described elsewhere [26, 71].
Dietary management
In patients with severe congenital malformations, no treatment is effective or appropriate. At the other end of the spectrum, some patients respond completely to riboflavin and CoQ10 and no dietary modification is necessary.
There are very few publications concerning dietary treatment. Designing an appropriate diet can be difficult and the degree of restriction needs to be tailored to the individual patient. Most patients with MADD are commenced on a low fat diet as the acyl-CoA dehydrogenases of β-oxidation are affected. Severely affected patients may also benefit from a modest protein restriction because MADD affects the breakdown of some amino acids (see above) in addition to fatty acids. Regular feeding with a high CHO intake is necessary to provide an adequate energy intake and to reduce lipolysis. Overnight fasting should be avoided in patients towards the severe end of the spectrum. A late night snack may be sufficient in some patients but others require a continuous overnight feed. Uncooked cornstarch could be substituted in older children to provide a ‘slow release’ source of glucose. It is essential to assess tolerance individually. It is advised that MCT should be avoided in MADD as it has appeared to have precipitated problems in some patients [72]. Medium chain fatty acids can enter the mitochondria independent of carnitine bypassing CPTI, the step at which β-oxidation is normally regulated. High concentrations of toxic metabolites may therefore be generated. At the author’s centre MCT has been given in small amounts to two patients, without any obvious problems.
The following provides a guide to the typical energy distribution of the diet in severe defects (based on personal practice):
- 65%–70% energy from CHO
- 8%–10% energy from protein (at least the safe level of protein intake for age [73])
- 20%–25% energy from fat (use of MCT is probably best avoided)
- frequent feeding
Some patients with severe MADD have deteriorated despite the above dietary measures. Oral treatment with the ketone body, sodium-D,L-3-hydroxybutyrate, has led to sustained clinical improvement in a few such patients [74]. It is important to take account of the sodium provided by this medicine when planning therapeutic feeds.
Monitoring
Regular monitoring of this complex diet is essential to ensure nutritional adequacy and normal growth. There is no useful biochemical marker to determine the response to diet. Routine biochemical nutritional monitoring should be undertaken for patients on low fat diets similar to LC-FAOD (p. 606) . If the child is on a very low protein diet, monitoring of plasma amino acids may be indicated.
Emergency regimen for use during intercurrent illness
During illness the standard ER of very frequent feeds, day and night, of glucose polymer should be given to minimise endogenous protein catabolism and lipolysis (p. 502) . These should be given via a tube if not managed orally. Fat should be avoided during the acute period. The normal diet can be introduced during the recovery period, but it is important to continue with frequent feeding and additional glucose polymer feeds until the normal diet is resumed.
Malonyl-CoA decarboxylase deficiency or malonic aciduria
Malonyl-CoA decarboxylase (MLYCD) catalyses the reaction of malonyl-CoA to form acetyl-CoA and regulates the intracellular concentration of malonyl-CoA, which is involved in the regulation of fatty acid oxidation through inhibition of CPTI [75]. Deficiency of MLYCD is recognised as having phenotypic overlap with LC-FAOD. It is a rare, autosomal recessive disorder with variable presentation and severity of developmental delay, seizures, cardiomyopathy (around 40% of cases), hypoglycaemia and acidosis [76]. The pathogenesis of all the clinical features seen in MLYCD deficiency have yet to be elucidated.
Biochemically there is raised malonylcarnitine in blood and increased urinary excretion of malonic and methylmalonic acids. Patients are treated with carnitine as total and free carnitine concentrations in plasma are low. Use of a low fat/high CHO diet and MCT supplements has been reported in individual cases as being of possible clinical benefit including the improvement of cardiomyopathy [76–79]. A biochemical response to diet has not been seen in all reported cases. There is limited detail and description of the diet for many of the published cases. The author’s centre reported improvement of cardiac function in a child when MCT provided around 25% of energy intake and LCT was minimal; she was also on an angiotensin-converting-enzyme (ACE) inhibitor [77]. MCT was given as these enter mitochondria directly, bypassing CPT1, and provide an alternative fuel for the heart. Prada et al. reported development of cardiomyopathy in a 5-month-old infant whose fat intake had been altered to provide 50% as LCT and 50% as MCT (combined fat intake 32 g). On altering the fat ratio further to 30% LCT and 70% MCT and commencing an ACE inhibitor her cardiac condition stabilised [78]. A trial of a low fat, MCT supplemented diet may be indicated in MLYCD deficiency patients who have cardiomyopathy. As yet there is inadequate evidence to know if diet may help in the prevention of onset of cardiomyopathy in patients diagnosed early or through expanded newborn screening. It is not yet known at the molecular level which patients may develop cardiomyopathy [76]. During illness the standard ER (p. 502) of very frequent feeds, day and night, of glucose polymer should be given to prevent metabolic decompensation.
Disorders of ketogenesis and ketone body utilisation
Ketones are an important fuel source which can be used by all tissues and particularly for the brain during prolonged fasting. They are also the preferred fuel for the heart. Ketones are formed from acetyl-CoA, produced mainly from the oxidation of fatty acids but also from the ketogenic amino acids leucine and isoleucine. Patients can present in the early neonatal period or later in childhood precipitated by prolonged fasting, intercurrent illness or other metabolic stress.
The disorders of biosynthesis of ketones (ketogenesis) and ketone body utilisation (ketolysis) include the following.
Defects of ketogenesis: 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) synthase deficiency and HMG-CoA lyase deficiency. The latter enzyme is also involved in the formation of ketones via the leucine degradation pathway (Fig. 19.2). The typical initial presenting symptoms include vomiting and lethargy which can progress to coma, with ‘hypoketotic’ hypoglycaemia, acidosis and hepatomegaly.
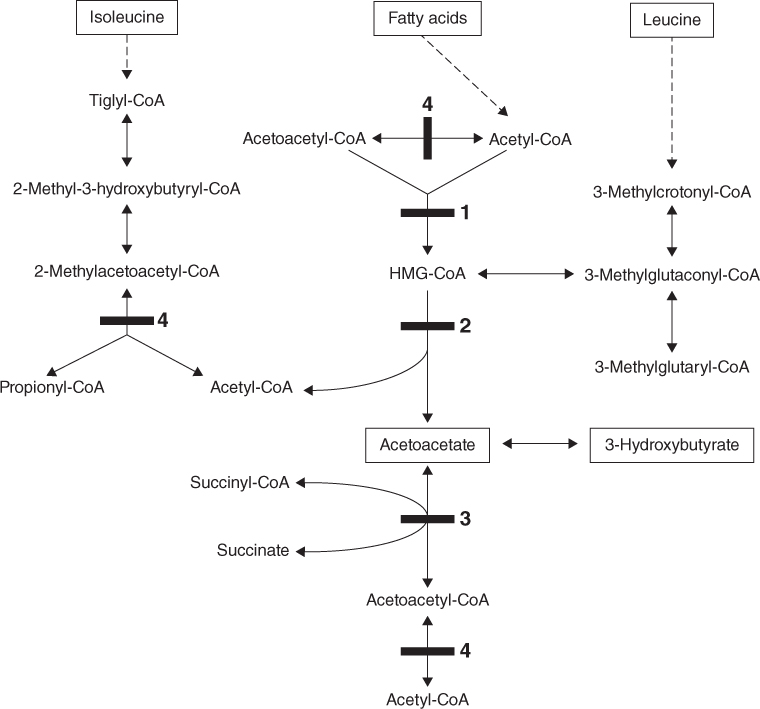
Figure 19.2 Biochemical pathways of ketogenesis and ketolysis. HMG-CoA, 3-hydroxy-3-methylglutaryl coenzyme A. 1, Mitochondrial(m) HMG-CoA synthase; 2, HMG-CoA lyase; 3, succinyl-CoA 3-oxoacid CoA transferase (SCOT); 4, mitochondrial acetoacetyl-CoA thiolase (T2).
Reprinted with permission of Springer.
Defects of ketolyis: succinyl-CoA 3-oxoacid-CoA transferase (SCOT) deficiency and 2-methylacetoacetyl-CoA thiolase (MAT) deficiency (commonly known as beta-ketothiolase deficiency, abbreviated to T2). The latter enzyme is also involved in the isoleucine degradation pathway (Fig. 19.2). Patients typically present with vomiting and high levels of ketones (as they cannot utilise them) which can cause a severe ketoacidosis, a reduced level of consciousness and dehydration.
A more detailed description of the clinical and biochemical features for these disorders can be found elsewhere [80, 81]. The disorders are all inherited as autosomal recessive traits.
Dietary management during illness
During illness the standard ER (p. 502) of very frequent feeds/drinks, day and night, of glucose polymer should be given to minimise lipolysis and endogenous protein catabolism (specifically the ketogenic amino acids).
In defects of ketogenesis provision of glucose will help prevent hypoglycaemia and limit the production of potentially toxic metabolites. Blood glucose monitoring by parents is not recommended as a marker of metabolic status, since hypoglycaemia is a relatively late finding and treatment should be initiated long before this develops. In defects of ketolysis, providing glucose will prevent illness from leading to ketoacidosis. Patients can become anorexic at an early stage, refusing ER drinks. It is essential feeds/drinks are therefore given via a nasogastric tube. In the event of persistent vomiting or the child not improving a hospital admission is needed for IV 10% glucose. Sodium bicarbonate may also be necessary to correct acidosis. Extra fluids will be needed if the child is dehydrated, which is common in the ketone body utilisation defects. Emergency management guidelines for use in hospital can be accessed at www.bimdg.org.uk.
As the child recovers and begins to eat again it is important that additional ER drinks of glucose polymer are given until the normal diet is fully resumed, particularly if they have not eaten well for several days. This needs to be managed carefully to prevent subsequent hypoglycaemia. If for any reason the child needs to be commenced on a special infant formula or paediatric feed during the recovery phase these should not contain added MCT which, once absorbed, are rapidly converted to ketones. In these disorders where there is a defect in either formation or utilisation of ketones, MCT may be of potential harm. Theoretically in SCOT and beta-ketothiolase deficiency MCT may increase concentrations of ketones (which cannot be utilised) and inhibit long chain fatty acid oxidation, thus contributing to the energy deficit. In the defects of formation of ketones, the conversion of MCT to ketones may be limited and so will not provide an energy source.
Dietary management – long term
There is little published information on the detailed dietary management of these disorders. Between episodes of metabolic decompensation patients are likely to be clinically asymptomatic. In the well child avoidance of prolonged fasts and provision of adequate energy is important. There are no published guidelines on safe fasting times for any of these disorders. In infants regular feeding, day and night, is important and it seems reasonable to continue a night time feed until the age of 1 year. Older children should tolerate overnight fasting (10–12 hours) without problems but they should be given a bedtime snack containing starch. Missed meals should be replaced with a high CHO drink if there is any risk of impending illness. MCT are theoretically best avoided as either they cannot be used to form ketones or they will add to the total ketone load which cannot be utilised.
A moderate protein (leucine) restriction (1–2 g protein/kg/day) is recommended for HMG-CoA lyase and beta-ketothiolase deficiency because these enzymes are involved in the catabolism of the ketogenic amino acids [82–84]. Also, in HMG-CoA lyase deficiency this has been combined with a modest fat restriction in some cases [82, 83]. However, opinions differ about the necessity of these dietary manipulations. Stable isotope studies suggest that protein catabolism contributes little to the abnormal metabolites in HMG-CoA lyase deficiency and fatty acid catabolism may be more important [85]. In the well child it is reasonable to suggest a generous CHO intake and avoidance of a high protein and fat intake.
Mild protein restriction (1.5–2.0 g/kg/day) has been described in SCOT deficiency (probably to limit increased concentrations of ketones being formed from the ketogenic amino acids) and avoidance of a fat rich diet which would cause ketosis [86, 87]. In the well child it is reasonable to suggest a generous CHO intake and avoidance of a high protein and fat intake.
A recent publication highlighted the potential foetal and maternal risks to women with HMG-CoA lyase deficiency, particularly the risk of metabolic decompensation associated with nausea and vomiting which needs careful management at an early stage [88].
Future research needs/unanswered questions
With the exception of MCAD deficiency, most disorders of fatty acid oxidation are rare and have only been described since the late 1980s; therefore long term experience of dietary treatment is limited and there are many unanswered questions about treatment. Moreover, in the long chain defects, treatment needs to prevent or delay long term complications such as retinopathy in long chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Some aspects of dietary treatment are the same worldwide; however, other aspects such as ‘safe fasting times’, use of overnight feeds and prescription for LCPUFA vary. Comparisons of different cohorts and further animal studies may begin to answer some of these questions. Proper evaluation of dietary treatment will require long term controlled studies in which other aspects of management are unchanged, but this can be very difficult to do.
Disorders of the Mitochondrial Respiratory Chain
Jacky Stafford
Introduction
These are a heterogeneous group of disorders characterised by impaired energy production. They are caused by mutations in either mitochondrial or nuclear DNA which directly or indirectly affect the oxidative phosphorylation (OXPHOS) system. The mode of inheritance is variable due to the large number of genes that encode respiratory chain proteins and ancillary factors: autosomal recessive, dominant, X-linked, maternal or sporadic. Mitochondrial disorders are estimated to affect 1 in 5000 children but this may be an underestimate [89].
Biochemistry
One of the major functions of mitochondria is the generation of ATP via the OXPHOS system which catalyses the oxidation of fuel molecules by oxygen into ATP, transferring electrons to oxygen via these complexes. Primary mitochondrial disorders are genetic disorders affecting any of the OXPHOS complexes, which are five functional enzyme complexes embedded in the inner mitochondrial membrane. Deficiency of any one of the enzymes will affect all cells containing mitochondria. Mitochondrial disorders may be classified by phenotype or enzyme deficiency which may be an isolated enzyme complex deficiency, e.g. commonly complex I or IV (cytochrome oxidase (COX) deficiency) or defects affecting several enzyme complexes [90, 91].
Disorders of OXPHOS affect the citric acid cycle due to excess NADH and lack of NAD. Ketone bodies increase particularly after meals due to channelling of acetyl-CoA towards ketogenesis. The blood lactate and lactate/pyruvate ratio is raised.
Clinical picture and treatment
Mitochondrial disorders may present at any age and certain clinical features are more frequent at certain ages. Complex I and IV deficiencies are more common (approximately 50%) among those disorders presenting in childhood [90]. Gastrointestinal symptoms and growth faltering are more often described as presenting features in infancy. Seizures may occur in 35%–60% of patients although clinical histories reveal they are preceded by other symptoms such as developmental delay and poor growth. Consistent factors are the increasing number of unrelated organs affected with time and the rapid progression involving the central nervous system at a later stage. The overlap of clinical features leads to difficulties in classification [92].
There is no satisfactory medical or dietetic therapy available; any treatment options are unproven and the prognosis is very poor [90, 93, 94].
Oral intake is poor and energy and/or nutrient dense supplements or supplementary feeds are used to support growth. Poor feeding is often associated with dysphagia and textural changes may be required. Continuous tube feeding may be necessary to manage gastrointestinal problems of nausea or early satiety (Table 19.9). A protein hydrolysate or amino acid based feed may be necessary if the child has diarrhoea. As the disorder progresses the feed may become the sole source of nutrition.
Table 19.9 Mitochondrial disorders whose syndromic presentations in infancy or childhood can require dietary management [89, 92]
| Name of disorder | Enzyme complex affected | Age of presentation | Symptoms and disorder specific treatment |
| Leigh syndrome (subacute necrotising encephalomyelopathy) | I, II, III, IV, V (singly or in combination); pyruvate dehydrogenase deficiency (PDH) is the most dominant phenotype (p. 614) [92] | Infancy (but can be later in childhood or occasionally in adulthood) | Vomiting, feeding difficulties Seizures; ketogenic diet tried but efficacy unclear [95] |
| Pearson syndrome | I, III, IV and V (all enzymes with mtDNA encoded subunits) | Infancy | Pancreatic insufficiency (requiring enzyme replacement therapy), diarrhoea, enteropathy Diabetes mellitus (may require insulin) |
| Alpers syndrome (progressive neuronal degeneration of childhood with liver disease) | I, III, IV and V (all enzymes with mtDNA encoded subunits) | Infancy | Bowel obstruction and perforation Seizures; ketogenic diet tried (one case report) but efficacy unclear [96] Liver failure (may be precipitated by sodium valproate) [89] |
| MELAS (mitochondrial encephalomyopathy, lactic acidosis and stroke like episodes) | I, III, IV and V (all enzymes with mtDNA encoded subunits) | Late in first decade | Anorexia, vomiting, poor feeding, enteropathy, abdominal discomfort, constipation, diarrhoea, gastroparesis, pseudo-obstruction and recurrent pancreatitis |
| Stroke like episodes may be due to decreased vasodilation capacity in small arteries which may be improved by supplementation with L-arginine [97] | |||
| MERRF (myoclonus epilepsy, ragged red fibres) | I, III, IV and V (all enzymes with mtDNA encoded subunits) | Childhood onset in ∼30% of cases | Rarely swallowing problems, gastrointestinal dysmotility [98] |
| MNGIE (mitochondrial neurogastrointestinal encephalopathy) | I, III, IV and V (all enzymes with mtDNA encoded subunits) | Generally between 10 and 30 years of age (average 18 years; can occasionally present in infancy) | Abdominal cramps, bloating due to gut dysmotility (dysphagia, nausea, vomiting, early satiety, abdominal pain, pseudo-obstruction and bloating) |
| Coenzyme Q10 deficiency (multisystem disease: nephropathy, encephalopathy including seizures, hearing loss) | Combined deficiency of I and III or II and III | Infancy (adult presentations include ataxia and myopathy) | Feeding difficulties and faltering growth related to nephropathy Response to replacement coenzyme Q10 to help restore electron transfer flow and antioxidant function is variable [99] |
| EME (ethylmalonic encephalopathy) | IV (COX) | Infancy | Diarrhoea |
Ketogenic diet in mitochondrial disorders
Ketone bodies can form ATP via the Krebs cycle and the OXPHOS system and may partially bypass complex I, thus potentially providing an alternative energy source for the brain and other tissues. In mitochondrial disorders a ketogenic diet (KD), high fat and low carbohydrate, may be beneficial to treat epilepsy in some specific subgroups. Complex I and IV deficiency represent a significant proportion of the group of patients with mitochondrial epilepsy: in a study of 48 children, 73% and 23% respectively [100]. KD was trialled in 24 of those children; 50% became seizure free and seizures were reduced by up to half in 25%. Kang et al. [95] reported safe and effective use of the classical KD in treatment of intractable epilepsy in 14 patients mostly complex I or IV, with seizure reduction (3/14) or seizure free (7/14). The same group describe better tolerance and adherence with use of the modified KD in 14 patients; 3/14 were seizure free and 5/14 had >50% seizure reduction [101]. However, it is unclear whether the KD affects disease progression in different mitochondrial subgroups and formal clinical trials are necessary [90, 95, 96, 101].
Pyruvate Dehydrogenase Deficiency
Fiona White
Introduction
Pyruvate dehydrogenase (PDH) deficiency is an inherited mitochondrial disorder of brain energy metabolism and is a major cause of primary lactic acidosis. The PDH enzyme is a complex comprising multiple copies of three subunits: pyruvate dehydrogenase (E1, consisting of two α and two β subunits), dihydrolipoamide (E2), dihydrolipamide transacetylase (E3) and E3 binding protein (E3BP). In addition there are two regulatory enzymes associated with the complex, PDH phosphatase and PDH kinase [102]. The majority of cases are due to mutations in PDHA1 which is an X-linked gene encoding the E1α subunit. A recent review of 371 patients reported 76% of cases resulted from deficits in the E1α subunit [103]. All other forms of PDH deficiency are autosomal recessively inherited [102] with the majority of cases having parents who are consanguineous, and often with several affected children in one family [103].
Biochemistry
The PDH complex is essential for the aerobic oxidation of glucose which is normally the obligate pathway for energy metabolism in the brain [102]. The PDH complex catalyses the rate limiting conversion of pyruvate, derived predominantly via glycolysis from glucose to acetyl-CoA. Pyruvate accumulates and is reduced to lactate in the cytoplasm. Pyruvate is in equilibrium with lactate and in PDH deficiency both blood and CSF lactate are elevated. The resulting lack of acetyl-CoA affects the availability of precursors feeding into the tricarboxylic acid (TCA) cycle and as a result limits mitochondrial energy production.
Clinical picture and treatment
PDH deficiency is a heterogeneous condition. The clinical spectrum is broad [103] ranging from severe neonatal presentation with overwhelming lactic acidosis, refractory to treatment and resulting in death in the neonatal period [104] to a more benign onset. Those with mild to moderate deficiency usually present in infancy with varied symptoms including developmental delay, seizures, hypotonia, failure to thrive, microcephaly, motor dysfunction [103, 105] and structural brain abnormalities as seen in Leigh syndrome [102].
In general, treatment is not very effective and prognosis is poor [106]. Sodium bicarbonate is used during periods of metabolic decompensation to manage lactic acidosis. Unlike in some other disorders, where lactic acidosis is a feature of metabolic decompensation and for which a high glucose emergency regimen may be used, this is not the case in PDH deficiency. A high glucose intake would result in further accumulation of pyruvate and lactate thus exacerbating the lactic acidosis. Chronically, a number of treatments are used although these rarely influence the course of the disease [104]. Supplementation with pharmacological doses of the cofactor thiamin is advocated as there is a rare cohort with the thiamin responsive form [104] where the mutation affects the interaction between thiamin pyrophosphate and the E1α subunit [103]. Dichloroacetate, a structural analogue of pyruvate, inhibits E1 kinase so improving activity of PDH complex and decreasing lactate levels and may benefit some patients [106]. Ketogenic diet (KD) treatment is employed in some cases as a means of providing an alternative energy source of acetyl-CoA, particularly for use by the nervous system [102]. Wexler et al. [107] reported the effectiveness of KD in E1 deficiency, improving cognitive development and survival. Response to KD therapy has also been reported by others in cases of E3BP deficiency [102] and E1 mutations [106]. Current practice in the author’s unit is to try KD treatment only in milder presenting patients, aiming to slow down the clinical progression. The outcome of KD treatment is variable with some patients having further metabolic decompensations and others either remaining static or making some developmental progress. A recent email survey of UK practice provided information on 22 patients on KD treatment; three were on MCT diet, 17 on classical diet and two on modified Atkins diet. Further details on the practical management of the KD, including monitoring, are given on p. 363.
Smith-Lemli-Opitz syndrome
Introduction
Smith-Lemli-Opitz syndrome syndrome (SLO) is an autosomal recessively inherited disorder of sterol biosynthesis caused by mutations on chromosome 11q13. More than 130 mutations have been described. The severe IVS8-1G>C is the most common among Caucasians (a third of reported cases) [108–110]. The incidence of SLO is reported to be 1 in 15 000 to 1 in 60 000, with highest prevalence in Caucasians [110].
SLO is due to a deficiency of 7-dehydrocholesterol reductase (DHCR7) which reduces the C7–C8 double bond in the sterol B ring, converting 7-dehydrocholesterol (7DHC) to form cholesterol, the final step of its biosynthesis.
Biochemistry
Patients with SLO have raised plasma 7DHC, the precursor of cholesterol. 8DHC, the isomer of 7DHC, is also raised and is probably synthesised from the accumulating 7DHC [110]. Plasma cholesterol is low or low to normal [111] but total cholesterol values may be in the normal range if they include significant amounts of 7DHC and 8DHC [109]. In SLO, the different fractions can be measured by specialist laboratories.
Clinical problems may be caused by the deficiency of cholesterol or total sterols, or the toxic effects of 7DHC and 8DHC and their derivatives, or a combination of all of these [108, 109]. Cholesterol is an important structural component of cellular membranes and myelin and a precursor of bile acids, steroid hormones, neuroactive steroids and oxysterols [109]. It has a role in signalling and defects in cholesterol metabolism affect the structure and function of the central nervous system. In the SLO affected pregnancy, mother to foetus cholesterol transport is limited and efficiency of transfer may affect severity of the disorder postnatally [108]. Clinical severity appears to correlate best either with absolute cholesterol levels or with the sum of 7DHC and 8DHC expressed as a fraction of total sterol [110].
Clinical picture and treatment
SLO is a multiple malformation syndrome with a wide phenotype ranging from a severe infantile presentation with early or perinatal death due to organ malformations, to minimal facial abnormalities and behavioural and learning difficulties [109].
Patients with SLO have a distinctive facial appearance, and typical craniofacial features include microcephaly, small upturned nose, ptosis, micrognathia, cleft palate (cleft lip is not common) or bifid uvula. Structural abnormalities are also observed: syndactyly of the second and third toes is the most common finding (95%) and in males genital abnormalities are frequently reported. In more severely affected patients there are often structural abnormalities of the heart, lungs, brain, limbs and kidneys and gastrointestinal tract, e.g. Hirschsprung’s disease, pancreatitis [108, 109].
Infants show growth faltering attributed to poor sucking and swallowing and a lack of interest in feeding [111]. They may also have psychomotor retardation, photosensitivity, behavioural abnormality, irritability, autistic tendencies and prefer not to be held [109]. An older child will demonstrate various degrees of hyperactivity, self-injurious behaviour, sleep disturbances and temperament deregulation [109].
As cholesterol is required for adrenal steroid and bile acid synthesis deficiencies of both may occur. The latter can cause a reduced ability to absorb cholesterol and fat soluble vitamins and bile acid supplementation may be required [100, 108] although a multicentre trial of 14 patients demonstrated no benefit of bile acid supplementation [112]. 7HDC and 8HDC may be monitored, but may have limited value for ongoing management.
Cholesterol supplements
A cholesterol supplement is given to raise plasma cholesterol. It may also decrease 7DHC and 8DHC synthesis via a feedback inhibition effect of plasma cholesterol on HMG-CoA reductase, diminishing endogenous cholesterol synthesis [108, 113]. Its effectiveness may be limited once normal cholesterol levels are achieved [114]. It also restores both adrenal and bile salt deficiencies [108].
Crystalline cholesterol in oil or aqueous based suspension and egg yolk cholesterol, in varying doses, has been reported to have different degrees of biochemical effectiveness [113, 115]. This may be due to the fact that cholesterol does not cross the blood–brain barrier and cannot treat the biochemical defect in brain [113]. Also, if 7DHC levels remain raised, detrimental effects may persist [109].
Clinically, the beneficial effects of cholesterol supplementation are not established as no controlled clinical trials have shown improvements in central nervous system function, i.e. behaviour and development [113]. A double blinded placebo crossover controlled trial showed no significant difference in short term behaviour over 2.5 months, using a placebo (egg substitute) or pasteurised egg yolk [116]. A standardised study of 14 patients indicated that cholesterol supplementation had little effect on developmental progress over a 6 year period [117]. There are anecdotal reports of better behaviour within days or weeks particularly in sleep disorders, irritability responding more quickly than other issues, e.g. tactile sensitivity [116], and improved growth with cholesterol supplementation [113]. It is also reported that behavioural abnormalities return when cholesterol therapy is interrupted [116]. It is generally considered that availability of cholesterol during foetal development is a major determinant in phenotypic expression of SLO. Most anomalies are of early embryonic origin and it is not feasible for treatment to cure patients postnatally [110].
HMG-CoA reductase inhibitor
Simvastatin reduces endogenous cholesterol synthesis by blocking the cholesterol biosynthesis pathway proximally at the first rate limiting step. It is proposed that this therapy may also reduce the levels of 7DHC and 8DHC in SLO. Simvastatin may also upregulate residual DHCR7 activity to increase cholesterol and decrease 7DHC and 8DHC [113]. Its use may be detrimental in subjects with little or no DHCR7 activity [114]. It can cross the blood–brain barrier and may have an effect on the central nervous system. A retrospective study of 39 patients showed no benefit with simvastatin on anthropometry or behaviour [114]. Clinical trials are needed to assess suitability of statins in SLO patients [109].
Nutritional support
Nutritional support is necessary in patients with postnatal growth failure and poor feeding [109, 111]. Growth charts for SLO have been produced from longitudinal data from 78 patients (0.1–16 years); 43% of patients were receiving treatment (nutritional support ± cholesterol) and showed growth restriction of 2 SD below norms for age [111].
Cholesterol supplementation of 60–100 mg/kg/day is given as divided doses twice or three times per day. It is preferred to a high cholesterol diet because achieving the recommended intake from diet would be difficult, particularly in those with poor oral intake. This may be administered via the feeding tube or taken orally. Different preparations are available.
Preparations used at the author’s centre include
- cholesterol suspended in Ora-Plus and flavoured with blackcurrant syrup (150 mg cholesterol/mL) (Cardiff and Vale NHS Trust). This product has a shelf life of 3 months and is stored in the fridge. It can be given orally or via a feeding tube.
- cholesterol powder (100 g tub) (Mawdsley Brooks Unlicensed Division). The powder is difficult to dissolve; in the past it was prepared in olive oil which had a very limited shelf life. It is now provided in powder form for addition to food or drinks.
Characteristics of patients with SLO are given in Table 19.10.
Table 19.10 Nutritional and gastrointestinal characteristics of 26 patients with SLO from three metabolic centres
| Metabolic centre | Number of patients | Number of patients requiring tube feeding | Reasons for tube feeding | Cleft palate | Number of patients requiring nutritional support | Number of patients with vomiting | Number of patients with constipation/loose stools |
| Birmingham Children’s Hospital NHS Foundation Trust, Birmingham | 7 (<17 years) | n = 2 (1 tube now removed) | Feeding difficulties | n = 3 high energy tube feeds or ONS n = 3 described as fussy eaters | n = 1 periodic vomiting but eats well | ||
| Central Manchester University Hospitals NHS Trust, Manchester | 7 | n = 1 with gastrostomy | Faltering growth, feeding difficulties | 1 | n = 4 sip feeds n = 2 poor appetite All described as fussy eaters | Most with constipation | |
| Great Ormond Street Hospital for Children NHS Foundation Trust, London | 12 | n = 4 with gastrostomy n = 8 orally fed (n = 2 had nasogastric tube previously) | Feeding difficulties: slow to feed, vomiting, slow to suck, not interested in feeding, spitting out food, growth faltering | n = 5 | n = 8 require high energy feeds and/or energy supplements (2) | n = 5 n = 2 required anti-reflux medicines n = 1 had Nissen fundoplication n = 3 had vomiting controlled by slow feed delivery | n = 4 n = 1 required lactulose n = 2 improved with solids n = 3 reported loose stools n = 2 improved with fibre containing feeds |
ONS, oral nutritional supplements.
Glycerol Kinase Deficiency
Introduction
Glycerol kinase deficiency (GKD) is an X-linked recessive disorder with mutations within or deletions on the GK gene in the Xp21 region. There is no genotype–phenotype correlation in isolated GKD [118]. Patients with complete deletions are symptomatic. Those with mutations resulting in residual GK enzyme function may be symptomatic or asymptomatic [119].
Glycerol kinase catalyses the phosphorylation of glycerol by ATP to yield glycerol-3-phosphate (G3P) and ADP. After phosphorylation 10%–30% G3P combines with free fatty acids to form triglycerides. The remainder is oxidised to dihydroxyacetone phosphate and enters the glycolytic or gluconeogenic pathway [120].
Biochemistry
GKD is characterised by hyperglycerolaemia and glyceroluria. Normal plasma glycerol levels (<0.2 mmol/L) may increase to 8 mmol/L resulting in urine levels >150 mmol/L (normally undetectable in urine) [120, 121]. In non fasting situations glycerol (via gluconeogenesis) contributes little to hepatic glucose production. In prolonged fasting conditions its contribution increases to about 20% energy as gluconeogenesis and fatty acid oxidation become essential for energy [122].
Clinical picture and treatment
There are three clinical forms based on phenotype [123]. Complex GKD is an Xp21 contiguous gene deletion syndrome involving GK locus together with congenital adrenal hypoplasia (CAH) and/or Duchenne muscular dystrophy loci. It usually presents neonatally and the clinical features depend on the loci involved [122]. Symptoms of isolated GKD may occur in addition to those of adrenal insufficiency, salt wasting, developmental delay and muscular dystrophy. Early diagnosis and prompt treatment to prevent hypernatraemic dehydration are required [122, 124, 125].
There are two forms resulting from isolated GK deficiency; the juvenile or benign adult form with pseudohypertriglyceridaemia [119, 123]. The juvenile form occurs in the first few years of life and is often precipitated by catabolism due to illness or poor feeding and can include episodic vomiting, metabolic acidosis, ketotic hypoglycaemia, lethargy, hypotonia, seizures and unconsciousness. Patients are at risk of insulin resistance, glucose intolerance and type 2 diabetes [118].
Avoidance of prolonged fasts is crucial [121, 123]. During illness an emergency regimen (p. 502) is necessary to prevent hypoglycaemia due to limited capacity to convert glycerol to glucose via gluconeogenesis. If IV fluids are required only glucose should be given and not lipids [123]. There are anecdotal reports that low fat diet (i.e. low in glycerol) may be beneficial for those experiencing symptoms of vomiting, acidaemia and stupor associated with GKD [126]. However, the mechanism for this is unclear. In the adult form, lipid lowering management is not reported to affect raised triglycerides [119]. At three main metabolic centres in the UK the standard emergency regimen during intercurrent illness is the only dietary treatment.
Barth Syndrome
Nicol Clayton
Introduction
Barth syndrome is an X-linked recessive genetic disease caused by defective remodelling of cardiolipin in the mitochondrial inner membrane due to mutations in the taffazzin (TAZ) gene [127]. Cardiolipin is the major phospholipid in mitochondria and has an important role in maintaining mitochondrial structure, the activity of the electron transport chain and other elements of the mitochondrial energy generating system [128]. The disorder was first described in 1983 [129] and the main characteristics are dilated cardiomyopathy, skeletal myopathy, growth retardation, neutropenia and 3-methylglutaconic (3-MGC) aciduria [130].
Epidemiology
Fewer than 200 living male patients have Barth syndrome worldwide and the Barth Syndrome Trust estimates the prevalence to be 1 in 300 000 to 1 in 400 000 live births. However, evidence is growing that the disease is significantly under-diagnosed [131].
Presentation
Boys with Barth syndrome typically present with symptoms of cardiac dysfunction or recurrent bacterial infections. Registry data show that 68% of patients develop cardiomyopathy in the first year of life [132]. Although neutropenia occurs in most patients with Barth syndrome, it is often intermittent and unpredictable and therefore might not be recognised when the cardiac disease is diagnosed. Increased concentrations of 3-MGC may be found by urine organic acid analysis. However, this is not a universal occurrence and levels can occasionally be normal [133], particularly in the newborn period. Other less specific presentations include faltering growth, feeding difficulties, biochemical disturbances such as hypoglycaemia or lactic acidosis (especially in neonates [134, 135]), episodic diarrhoea and delayed motor milestones; there are also characteristic facial features with a cherubic appearance (chubby cheeks and deep-set eyes) and prominent ears [136].
Diagnosis
Confirmation of a clinical diagnosis of Barth syndrome can be made either by mutation testing of the TAZ gene or by cardiolipin analysis using whole blood or stored blood or tissue, such as Guthrie cards or skin fibroblasts. Abnormal results of cardiolipin testing are followed up with mutation analysis to confirm the genetic diagnosis. Diagnostic testing by cardiolipin analysis is free of charge at the NHS Specialised Services Barth Syndrome Service, Bristol Royal Hospital for Children (see Useful addresses). Due to the variable nature of the neutropenia and levels of 3-MGC, all boys with idiopathic dilated cardiomyopathy should be screened for Barth syndrome.
Medical treatment
Historically, most boys with Barth syndrome died during infancy due to either heart failure or sepsis. However, advances in early diagnosis and treatment have reduced mortality significantly, although there is evidence that Barth syndrome is a cause of miscarriage and stillbirth [137, 138]. After diagnosis, patients are treated for their cardiac failure and given prophylactic antibiotics and granulocyte colony-stimulating factor (G-CSF) when significant neutropenia is present. The Bristol NHS Specialised Services Barth Syndrome Service runs bi-annual clinics and offers multidisciplinary care from specialists in haematology, cardiology, psychology, nursing, dietetics, genetic counselling, physiotherapy, speech therapy and occupational therapy.
Biochemistry
Cardiolipin deficiency alters the functioning of proteins within mitochondria [139]. Ongoing studies on the intermediary metabolism of Barth syndrome suggest that abnormalities of the cardiolipin-deficient, inner mitochondrial membrane lead to a disordered flow of substrates through the citric acid – or tricarboxylic acid (TCA) – cycle. This in turn reduces the complete oxidation of acetyl-CoA from the breakdown of glucose and fat and increases the catabolism of those glucogenic amino acids that enter the TCA cycle at or after alpha-ketoglutarate, a process termed ‘anaplerosis’. The diversion of specific amino acids is thought to be sufficiently great to lower plasma amino acid levels and impair muscle protein synthesis. Thus, whereas there is sufficient anaplerosis to maintain normal or near normal energy metabolism, this occurs at the cost of wasting of muscle, potentially including cardiac muscle. Of particular interest are the amino acids arginine and cysteine, as children with Barth syndrome often have fasting levels in the lower range of normal [140].
Growth delay
Growth delay is common in Barth patients [141] and the growth pattern is consistent with constitutional growth delay with late onset puberty. Delayed bone age, with a range between 8 months and 2 years 6 months, has been seen in all boys attending the author’s Barth syndrome clinic. Reduced nutritional intake due to cardiomyopathy, sore mouth/gums and recurrent infections may also contribute to poor weight gain.
Boys with Barth syndrome are typically normal size at birth, but their growth rate progressively decelerates in the first 2–3 years. Height usually falls to the 2nd centile, and often well below the 0.4th centile, by the third year with a concurrent deceleration in rates of weight gain. Until onset of a late puberty, height remains on or below this centile, although the trajectory of growth remains parallel to the centile. Because of the almost universal muscle hypoplasia seen in Barth syndrome, weight centile can be 1 to 2 standard deviations below height centile. Using data from the Barth syndrome registry, Spencer et al. [142] showed that 58% of patients under 18 years of age were 5th centile for weight and at or below the ≤5th centile for height. However, once boys reach puberty at 15–18 years of age, they grow rapidly, achieving their predicted adult height by their late teens or early 20s. The average centile reached after 18 years is the 50th centile, although some boys exceed this and reach above their mid-parental height centile.
In line with constitutional growth delay, mildly to moderately depressed levels of insulin-like growth factor 1 (IGF-1) and growth hormone may be found in Barth syndrome. Because most boys have bone ages in accordance with height age and low growth velocities, the low hormone levels are likely to be a reflection of the slow growth pattern rather than a sign of an underlying endocrinopathy.
Not all children with Barth syndrome display poor weight gain. Although just over half of boys have a weight <5th centile, nearly 25% of boys younger than 18 years have a weight >20th centile and more than 50% have normal or increased body mass index (BMI) [142]. Data from the Bristol UK clinic show an increasing trend of boys becoming overweight for their height and body composition measurements, using dual X-ray absorptiometry and bioelectrical impedance, and show that even when weight and height are on similar centiles the boys have higher body fat percentages and lower muscle mass. As a result, an appropriate healthy BMI in Barth syndrome will be at or below the 25th centile for age. Boys with BMI >25th centile have markedly abnormal, often truncal, adiposity, possibly aggravated by misconceived overfeeding to achieve normal weights for age (unpublished observations). However, in view of the marked deficiency of oxidative metabolism in muscle, where most dietary fat is metabolised, a propensity to excess adiposity is to be expected and needs to be addressed in the dietary management of Barth syndrome.
Dietary management
There are few published data on nutritional requirements. Dietary management is individualised, based on maintaining growth within appropriate parameters. The key nutritional aims are to
- provide adequate but not excessive energy to maintain appropriate growth and keep the weight centile within 2 centiles of the height centile. Energy requirements start to decrease after infancy and by the age of 5 years are likely to be around 70% of the estimated average requirement (EAR) for chronological age depending on activity.
- provide sufficient protein to promote muscle protein synthesis and mitochondrial anaplerosis with an even and regular distribution of protein throughout the day. A higher than usual percentage of energy as protein should be given with a protein intake up to 2 g/kg/day.
- limit fasting to reduce muscle proteolysis, thereby preserving muscle mass. A regular intake of low glycaemic index (GI) carbohydrates should be given throughout the day with cornstarch therapy or a carbohydrate rich snack before bed (p. 622) .
- encourage low fat food options, particularly dairy products and between meal snacks, if the weight centile is at or above the height centile.
- supplement vitamin and mineral intake to ensure 100% of the reference nutrient intake (RNI) is achieved, particularly with respect to vitamin B6.
Infancy
Infants with Barth syndrome often have difficulty feeding and maintaining sufficient feed volumes to allow for growth. This is due in part to low muscle tone and early fatigue with feeding, but may also be secondary to cardiac dysfunction in more severe cases. Infants typically take very small amounts and feed frequently throughout the day and night, often feeding during the night until after their second year.
Whereas energy requirements during the first 6 months are usually normal for weight and chronological age, energy requirements reduce to around 90% of the EAR as growth slows towards the end of the first year. Nutrition should be targeted at maintaining an appropriate weight velocity, with a weight centile that is likely to be within one to two centiles of the height centile. Protein requirements are increased in Barth syndrome and unless a high protein diet is otherwise contraindicated, the aim is to give up to 2 g/kg/day throughout childhood.
Although a small number of infants with Barth syndrome have successfully breast fed, due to the low volumes taken and the need to ensure an adequate protein intake, supplemental feeding with high energy formulas or nasogastric feeding can be necessary. However, tube feeding can cause infants to become rapidly overweight and have a negative impact on oral feeding, so regular monitoring is required. Caution should also be exercised if a high target for energy intake, e.g. 120–150 kcal (500–630 kJ)/kg, is used in an attempt to achieve catch up weight after a period of faltering growth as this can cause vomiting and exacerbate diarrhoea.
Hypoglycaemia and (usually mild) lactic acidosis has been reported, particularly in the first year, although this usually resolves after infancy. However, unresponsive hypoglycaemia following 24 hours of fasting during a period of acute illness has been seen in a 2 year old (personal observation). The reasons for hypoglycaemia in Barth syndrome are not fully understood and families are encouraged to ensure that infants do not go for long periods without feeds; early admission to hospital is advised if intercurrent illness substantially limits feeding.
Weaning can commence at the normal time around 6 months of age, although progression onto a full diet is often slow paced and parents should expect a marked delay, with infants at 1 year still depending on regular small milk feeds and tolerating only smooth textures. As many Barth infants have poor muscle tone, care must be taken to ensure that they are able to sit up and support their heads in order to reduce the risk of choking. Gagging is commonplace and this often precipitates vomiting. Moving onto stronger tasting food and encouraging bite and dissolve finger foods when safe to do so can help make weaning more enjoyable. Delayed physical maturation and often repeated hospitalisations during infancy can also compound weaning being delayed and critical developmental stages being missed. If transition to solid food is not possible at 6 months, families should be encouraged to offer very small amounts of foods, such as puréed fruits and vegetables, yoghurts and cheese sauces, given from finger tips or the tip of a spoon to expose the infant to a variety of tastes until full weaning can commence.
Childhood
Many boys continue their slow transition to solid food during the first 3 years and will continue to have a degree of oromotor weakness and sensory issues. A sensitive gag reflex and poor chewing skills can lead to extended mealtimes and small portion sizes should be expected. Some boys continue to receive supplemental feeding and moving onto a solid diet can be problematic, especially when excessive energy is being given by tube feeding or supplement drinks. A slow approach is the most successful including sensory play with food (cooking, tasting, playing), involving the boys in mealtimes (particularly with their peers) and being positive and encouraging about their preferred food choices, which may be unusual (see below). The amount of tube feeds or oral nutritional support should be reviewed frequently to ensure that appetite for food is not being suppressed by excessive energy intake from feeds.
Restricted variety in the diet is commonplace and boys favour milk, cheese, crisps and toast (Bristol Clinic data). There is a propensity for food fads, especially for unusual savoury foods. A patient list-serve email discussion on food fads (December 2011) described boys having cravings for pickles, hot/chilli sauce, olives, mayonnaise, ketchup, soy sauce, cheese, milk and especially salt. Behaviour ranged from adding salt to food through to drinking the brine from feta cheese to eating salt by itself. There is no current evidence for a salt wasting state or any other explanation for this almost universal craving for salt. Judicious use of salty tasting sauces and dips when offering new foods can help encourage boys to expand the range of foods that they will eat. Limiting high fat foods, such as crisps and full fat dairy products, is sensible if they are overweight.
Energy requirements reduce throughout childhood as a consequence of reduced physical activity, slower growth and a lower muscle mass. By the age of 5 years, energy requirements can drop to 70% of the EAR so if boys are tube fed their weight gain must be carefully monitored to make sure they are not overfed. Protein requirements remain high at up to 2 g/kg/day and additional protein, vitamin and mineral supplements may be required if feed volumes are low.
Children who do eat are encouraged to have regular small meals and snacks and where possible, each containing a source of protein and carbohydrate. The diet should start to contain more complex low GI carbohydrates such as multigrain bread, pulses, wholegrain cereals and oats. A lower fat diet should be encouraged for those whose weight is on a higher centile than their height; for all other boys fat intake should be in line with current UK guidelines for children over 5 years of age, i.e. no more than 35% of total dietary energy. As most boys have low arginine and cysteine levels, foods with a higher content of these amino acids (such as nuts, lentils, sunflower seeds, tuna, chicken, salmon, prawns, dairy products and eggs) may be helpful.
Adolescence
Boys with Barth syndrome have delayed puberty, typically with onset between 15 and 18 years of age. When this later pubertal growth spurt does occur it is often without a significant increase in food intake, with the result that many previously slim boys may become abnormally thin. This, together with a worsening of cardiac function to some degree during puberty, may indicate a need for supplemental feeding via nasogastric or gastrostomy tubes.
Vitamins and minerals
Vitamin and mineral requirements are often not met due to small portion sizes, restricted range of foods taken and lower energy requirements. There is a potential increased need for vitamin B6 due to increased amino acid catabolism; low functional levels of B6 may contribute to poor plasma cysteine levels, although serum B6 levels in all boys tested at the author’s clinic were within the normal range. Evidence of B6 insufficiency has been seen as a block in the transulfuration pathway, manifested as inappropriately high levels of plasma methionine that normalise immediately with B6 supplementation (unpublished observations) and further investigation of this is under way. A number of the boys have low vitamin A levels which do not respond to supplementation; toxicity may occur with continued high doses. Vitamin D levels can be low, although the trends and percentage of boys who have suboptimal vitamin D status are the same as those seen in the general population (UK Bristol Clinic data 2010–2013). To ensure an adequate intake of all vitamins, it is recommended that boys taking diet alone have a multivitamin and mineral supplement that provides up to 50% of the RNI. Those receiving oral nutrition supplements (e.g. Paediasure, Fortini, Ensure) which are fortified with vitamins and minerals may require a lower dose.
Carnitine and coenzyme Q10
A number of mitochondrial cocktails have been tried in Barth syndrome, including giving L-carnitine [143] and pantothenic acid [144]. With the exception of one isolated success, these results have not been favourable [145]. Although some benefit from treating carnitine deficiency (total plasma carnitine level <20 µmol/L) is possible in Barth syndrome, any high dose carnitine treatment (e.g. 50–100 mg/kg/day) given in the absence of deficiency, as used in some metabolic disorders, can precipitate cardiac failure and muscle weakness and should be avoided (patient registry, Barth Syndrome Foundation, 2011). There is no evidence that coenzyme Q10 is beneficial in Barth syndrome.
Omega-3 fatty acids
The anti-arrhythymic effects of fish oils has led to the suggestion that supplementation could be beneficial in Barth syndrome. Early work has established that neutrophil counts are not altered by docosahexaenoic acid (DHA) supplementation (personal communication). Until more investigations are undertaken, current recommendations are to have two portions of fish per week, one of which should be oily fish, in line with current UK guidelines [146].
Cornstarch therapy
During an overnight fast, degradation of muscle and other body protein occurs to release amino acids for gluconeogenesis when liver glycogen stores have been exhausted. This small muscle loss can be significant in Barth syndrome, where muscle mass is already reduced and the ability to rebuild it during the daytime is diminished due to hypotonia and decreased physical activity. Supplementary cornstarch at bedtime provides a slow release of glucose overnight and is a protein sparing treatment aimed at reducing overnight fasting time.
Cornstarch therapy can be started once overnight feeding has stopped at about the age of 2–3 years. The dose is built up to a maximum of 2 g/kg (as tolerated) and mixed into milk or yoghurt and given before bed. If cornstarch therapy is rejected, it is beneficial to offer a small snack close to bedtime, ideally containing protein and low GI complex carbohydrate, such as yoghurt or milk with a small oat biscuit or wholegrain cracker, multigrain toast or cereal.
Dietary management of illness
Careful attention is necessary during any intercurrent illness especially where there is a reduced or no intake of feed or food, such as vomiting. During this time children should be given an oral rehydration solution (p. 106) supplemented with glucose or glucose polymer up to 10% carbohydrate concentration. This should be offered frequently throughout the day to reduce the risk of hypoglycaemia. Drinks and emergency regimens containing higher concentrations of glucose, and extended periods of receiving carbohydrates only, should be avoided as the effect is to lower amino acid levels which may depress TCA cycle activity further. A source of protein should be introduced as soon as tolerated.
If fasting exceeds 24 hours, or the child is less than 1 year old, hospital admission is recommended for early treatment with intravenous (IV) glucose at 10% concentration. If feeding cannot be restarted within 48 hours of admission, and consequently protein intake is minimal, enteral or parenteral nutrition should be considered.
Diarrhoea
Diarrhoea may occur as a persistent or intermittent feature; it is not clear whether this is caused by viruses or abnormal smooth muscle function. Although most children with Barth syndrome do not have fat malabsorption this has been occasionally reported.
A possible contribution to diarrhoea is overproduction of acetylcholine, a neurotransmitter that increases contractility of smooth muscle in the intestine, via shunting of citrate (a TCA cycle intermediate and precursor of acetylcholine) out of the TCA cycle (personal communication). Citric acid is found naturally in many fruits and vegetables, and is most concentrated in oranges, lemons, lime, grapefruit, berries and tomatoes. Some cheeses (especially mozzarella) contain citric acid and it is added to foods (as the preservative E330) in particular to soft drinks, jams and preserves, fruit yoghurts, tinned tomatoes, sweets, sherbet, some ice creams and some flavoured crisps. Although there is no evidence that citrate or citric acid should be completely excluded from the diet, some patients have experienced improvement in symptoms when citric acid is reduced; therefore a trial reduction may be helpful.
During prolonged diarrhoea low muscle mass, and therefore lower potassium stores, can result in a more rapid depletion of potassium so electrolytes should be closely monitored. Care is also needed with IV fluids containing potassium, as patients may become hyperkalemic more quickly than would be expected.
Ongoing work
Specific amino acid supplementation
A number of boys in the USA, Europe and Australia have received amino acid supplementation with combinations of arginine, citrulline, cysteine and methionine to normalise fasting blood levels of these amino acids and in turn enhance muscle protein synthesis. The amounts and combinations are individually titrated. This work is being undertaken by Dr Richard Kelley, Professor of Paediatrics, Johns Hopkins School of Medicine, Baltimore, USA. Evidence based guidelines for amino acid supplementation will be produced from this work. Early reports include improvements in weight gain, appetite and cardiac function. There is ongoing work into the efficacy, tolerance and safety of this treatment.
Lipid Disorders
Janine Gallagher
Introduction
There are three main pathways responsible for the production and transport of lipids in the body: the exogenous pathway; the endogenous pathway; and the reverse cholesterol transport (Fig. 19.3 and Table 19.11). There are three main types of lipids: triglycerides, cholesterol and phospholipids. Lipids are transported in plasma as lipoproteins; these have a core of hydrophobic lipids (triglycerides and cholesterol esters) with amphiphilic molecules on the surface (phospholipids, cholesterol and proteins known as apolipoproteins). There are four main classes of lipoproteins (Table 19.11) which are categorised according to their density.
Figure 19.3 Lipid transport [147].
Cm, chylomicron particles; CmR, chylomicron remnant particles; VLDL, very low density lipoproteins; HDL, high density lipoproteins; LDL, low density lipoproteins; IDL, intermediate density lipoproteins; CETP, cholesterol ester transfer protein; FFA, non esterified fatty acids; LCAT, lecithin-cholesterol acyltransferase; Apo, apolipoprotein.
Reprinted with permission of Oxford University Press.
Table 19.11 Lipoprotein composition and function [148]
| Lipoprotein | %wt TG | %wt PL | %wt C+CE | Apoproteins | Function |
| Cm | 90 | 5 | 4 | Apo B-48, C-I, C-II, Apo A-I, A-IV, C-III, E | Transport of absorbed TG from intestines and hydrolysed by endothelium-bound LPL in adipose tissue and muscle. CmR taken up by liver (exogenous pathway) |
| VLDL | 55 | 19 | 20 | Apo B-100, C-I, C-II, C-III, Apo A-I, A-II, E | Transport of TG synthesised in liver to adipose tissue and muscle (endogenous pathway) |
| LDL | 5 | 20 | 55 | Apo B-100, Apo C-I, C-II, C-III, E | Transport of cholesterol from liver to peripheral tissues (endogenous pathway) |
| HDL | 5 | 27 | 21 | Apo A-I, A-II, Apo C-I, C-II, C-III, E | Transport of cholesterol from peripheral tissues to liver (reverse cholesterol transport) |
Cm, chylomicrons; CmR, chylomicron remnants; VLDL, very low density lipoproteins; HDL, high density lipoproteins; LDL, low density lipoproteins; TG, triglyceride; PL, phospholipid; C+CE, sum of cholesterol and cholesterol ester; LPL, lipoprotein lipase.
Disorders of lipid metabolism which are managed by dietary intervention can be classified into those in which the serum lipoproteins are deficient or absent, hypolipoproteinaemia, or in which they are increased, hyperlipoproteinaemia.
Hypolipoproteinaemias
Abetalipoproteinaemia, hypobetalipoproteinaemia and chylomicron retention disease are rare genetic diseases which cause malnutrition, growth failure and vitamin E deficiency [149].
Abetalipoproteinaemia
Abetalipoproteinaemia (ABL) is an autosomal recessive disorder, incidence <1 in 100 000, caused by mutations in the microsomal triglyceride transfer protein (MTP) gene resulting in the absence of plasma apolipoproteins (apo), B-containing lipoproteins, chylomicrons, VLDL and LDL [150]. MTP facilitates the transfer of lipids onto apo B. Presenting features are given in Table 19.12.
Table 19.12 Presenting features of abetalipoproteinaemia
| Diarrhoea, steatorrhoea |
| Faltering growth due to fat malabsorption |
| Fat soluble vitamin deficiency (A, D, E, K) |
| Low plasma lipids and apolipoprotein B levels |
| Low plasma levels of essential fatty acids |
| Liver function tests may reveal elevated serum transaminases with hepatomegaly due to hepatic steatosis [151] |
| Acanthocytosis |
| Anaemia due to iron and folate deficiencies secondary to fat malabsorption [152] |
| Elevated prothrombin time and in severe cases gastrointestinal bleeding associated with vitamin K deficiencies [151] |
| Ophthalmic involvement, predominantly retinitis pigmentosa |
| Neurological manifestations including areflexia, ataxia, dysarthria and possibly cognitive decline |
| Muscle involvement including myopathy |
The onset of neurological symptoms usually begins in the first or second decade of life [153]. Prior to the use of high dose oral fat soluble vitamin treatment, neurological complications developed before the second decade and in some cases survival was not past the third decade [154]. Ophthalmic involvement is variable and covers a wide range of symptoms and manifestations [155]. The neurological and ophthalmic manifestations appear to be secondary to deficiency of vitamin E; vitamin A deficiency may also contribute to retinopathy.
Dietary management
Treatment involves reduction of dietary fat to prevent steatorrhoea and supplementation with tocopherol to prevent progression of the neuromuscular and retinal degenerative disease [156]. Fat restriction is determined by individual tolerance (monitoring gastrointestinal, GI, symptoms) which increases with age and can vary from 5 g fat/day in infants up to 20 g/day in the older child. In the author’s centre fat intake has been maintained at 10 g/day, not inclusive of fat derived from walnut oil, which is given to provide essential fatty acids (EFA). Despite the inability to secrete chylomicrons, there does appear to be some absorption of long chain fatty acids [157]. These are probably transferred to the liver as free fatty acids. Dietary treatment should aim to alleviate GI symptoms and increase energy intake to allow for catch up growth.
Medium chain triglycerides
The fatty acids derived from medium chain triglycerides (MCT) (C:8, C10:0, C12:0) do not require chylomicrons for their absorption as they are transported directly from the intestine to the liver via the hepatic portal system. The use of MCT as an energy source in ABL is controversial. There have been several reports of hepatic fibrosis with the use of MCT [158, 159], but there have also been reports of children who developed cirrhosis independent of the use of MCT oil [160, 161]. There are case reports of the benefit of MCT [162] and absence of adverse effects with its use [163]. Historically, MCT has not been used in the treatment of ABL, but some metabolic centres have started to use it; regular liver ultrasound scans and biochemical monitoring of liver function is required.
Infant feeds
An example of a minimal fat modular feed is outlined in Table 19.13. The feed requires a minimal fat protein source (e.g. Protifar, Vitapro, skimmed milk powder), a carbohydrate source as glucose polymer (e.g. Polycal, Vitajoule), electrolytes, a source of EFA (e.g. walnut oil), vitamins and minerals (e.g. Paediatric Seravit). Long chain triglycerides (LCT), given as a fat emulsion, e.g. Calogen, are added gradually to the feed as tolerated. LCPUFA can be given as KeyOmega. Consideration could be given to the use of Walnut oil as a LCT source if EFA remain low.
Table 19.13 Minimal fat feed (<5 g fat) for use in abetalipoproteinaemia
| 16-week-old boy, prior to weaning, weight 5.4 kg (2nd centile), length 60 cm (9th centile) | ||||||
| Daily requirements: | ||||||
| fluid 150 mL/kg, energy 95 kcal (400 kJ) to 130 kcal (545 kJ)/kg, protein 1.6 g to 3 g/kg | ||||||
| EFA given as walnut oil to provide 0.1 mL/56 kcal (235 kJ) of the estimated average requirement (EAR) for energy for age [57] | ||||||
| This can be given as 180 mL × 6 feeds | ||||||
| Energy (kcal) | Protein (g) | CHO (g) | LCT (g) | Na+ (mmol) | K+ (mmol) | |
| Vitapro 15 g | 54 (225 kJ) | 11.3 | 1.4 | 0.9 | 2.0 | 2.7 |
| Polycal 115 g | 442 (1850 kJ) | – | 110.4 | – | 0.2 | 0.1 |
| Paediatric Seravit 15 g | 45 (190 kJ) | – | 11.3 | – | 0.1 | – |
| Calogen 6 mL | 27 (115 kJ) | – | – | 3.0 | – | – |
| Walnut oil 1 mL | 8.4 (35 kJ) | – | – | 0.9 | – | – |
| KCl 6 mL (2 mmol/mL) | – | – | – | – | – | 12.0 |
| NaCl 1.5 mL (5 mmol/mL) | – | – | – | – | 7.5 | – |
| Water to 840 mL | ||||||
| Total | 576 (2415 kJ) | 11.3 | 123.1 | 4.8 | 9.8 | 14.8 |
| Per 100 mL | 69 (285 kJ) | 1.3 | 14.6 | 0.6 | 1.2 | 1.8 |
| Per kg | 107 (445 kJ) | 2.1 | 22.8 | 0.9 | 1.8 | 2.7 |
An extremely low fat infant formula, Basic-f (Milupa), is available providing, per 100 mL (13% dilution), 49 kcal (207 kJ), 10.2 g CHO, 1.8 g protein and <0.1 g fat (p. 597). A source of EFA must be added together with LCT (to tolerance) and glucose polymers to provide adequate energy.
Historically breast feeding has been considered to be contraindicated in ABL; however, partial breast feeding may be possible [164] in combination with a minimal fat feed. Close monitoring is required and the ratio of breast feeding to formula feeding altered according to biochemical parameters, clinical symptoms and growth.
Weaning
At the usual time for weaning, around 26 weeks of age, minimal fat foods can be commenced, e.g. baby rice mixed with the minimal fat feed, fruits and vegetables (except avocado pears and olives), pulses, meats and fish naturally low in fat (Table 19.14). As a guide for using commercial baby foods, wet baby foods and dried baby foods containing ≤0.5 g fat/100 g and ≤2 g fat/100 g, respectively, are allowed freely if given in usual portion sizes. Other baby foods with a higher fat content would need to be counted as part of the total daily fat allowance.
Table 19.14 A sample minimal fat weaning menu
| On waking | Minimal fat feed |
| Breakfast | Suitable commercial baby cereal* mixed with minimal fat feed |
| Mid-morning | Minimal fat feed |
| Lunch | Purée turkey, chicken (white only), white fish or lentils |
| Purée or mashed potato/sweet potato/pasta/rice (white) | |
| Purée or mashed vegetables | |
| Suitable savoury commercial baby food* | |
| Purée fruit or suitable commercial baby fruit dessert* | |
| Very low fat yoghurt/fromage frais | |
| Milk pudding (custard, ground rice) made with minimal fat feed or skimmed milk | |
| Mid-afternoon | Minimal fat feed |
| Evening meal | As lunch |
| Bedtime | Minimal fat feed |
| Extra feeds or water can be given with meals | |
| Suitable finger foods from 6–9 months: fingers of toast (white bread)* (no spreading fat), soft cooked broccoli florets/carrots, soft pear/mango, banana slices | |
* Check fat content of product.
Families can calculate the fat content of baby foods from nutrition labels so that higher fat foods can be counted in the total daily fat intake.
To calculate 1 g fat from a food product:
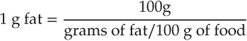
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


