Chapter Outline
ACQUISITION AND DISTRIBUTION OF IRON
ROLE OF IRON IN CELL PROLIFERATION AND DIFFERENTIATION
REGULATION OF PROTEINS OF IRON METABOLISM
Copper Physiology and Transport
Hematologic Aspects of Copper Deficiency
Hematologic Aspects of Acute Copper Intoxication and Wilson Disease
Iron lacks the glitter of gold and the sparkle of silver but outshines both in biologic importance. This plebeian metal is vital to the function of a wide variety of critical enzymes, including catalases, aconitases, ribonucleotide reductase, peroxidases, and cytochromes, which exploit the flexible redox chemistry of iron to execute a number of chemical reactions essential for our survival ( Box 11-1 ). In addition, we depend on hemoglobin, another iron-containing protein, to transport inhaled oxygen from the lungs to peripheral tissues. Human existence is inextricably linked to iron, and disturbances in its metabolism may have dire consequences.
HEME PROTEINS
Hemoglobin
Myoglobin
Cytochrome a , b , c
Cytochrome P-450
Tryptophan 1,2-dioxygenase
Catalase
Myeloperoxidase
IRON-DEPENDENT ENZYMES
Aldehyde oxidase
Reduced nicotinamide adenine dinucleotide dehydrogenase
Tyrosine hydroxylase
Succinate dehydrogenase
Prolyl hydroxylase
Tryptophan hydrolase
Xanthine oxidase
Ribonucleotide reductase
Aconitase
Phosphoenolpyruvate carboxykinase
Physiologic Chemistry of Iron
Iron and Oxidation
The key to the biologic utility of iron is its ability to exist in either of two stable oxidation states: Fe 2+ (ferrous) or Fe 3+ (ferric). This property permits iron to act as a redox catalyst by reversibly donating or accepting electrons. An excellent example is the electron transport chain of oxidative phosphorylation, in which adenosine triphosphate (ATP) is generated from glucose by the orderly transfer of electrons through a network of iron-containing mitochondrial cytochromes.
When dissolved in aqueous solution, ferrous iron rapidly oxidizes to ferric iron, which is insoluble at physiologic pH. The resulting ferric hydroxide salts (rust) are of no metabolic utility. To achieve stable solubility under physiologic conditions, iron complexes with iron-binding agents termed chelators. Chelators are synthesized by all organisms, ranging from microbes (e.g., deferoxamine produced by Streptomyces pilosus ) to humans (e.g., transferrin in human plasma). These molecules are important to environmental iron acquisition and transport as well as intracellular storage.
Iron-Protein Complexes
Iron-protein complexes capitalize on the properties of the metal to perform metabolic functions, the iron acting as the chemical workhorse and the protein dictating biologic specificity. Individual iron atoms can interact directly with amino acid side groups in proteins, as in ribonucleotide reductase. Alternatively, iron may form coordination complexes with other small molecules. For example, protoporphyrin IX donates four of the six electrons needed to form a stable coordination complex with iron. Heme, the iron–protoporphyrin IX complex, is so stable that removal of the iron moiety requires an enzyme, heme oxygenase. The functional properties of heme are determined by the nature of the associated protein or small molecule ligands supplying the remaining two electrons. The best-characterized heme protein is hemoglobin, in which a globin histidine residue donates the fifth electron and the sixth comes from molecular oxygen. This configuration enables hemoglobin to transport oxygen safely throughout the body.
Iron and sulfur atoms can also form complexes (clusters) that catalyze enzymatic reactions. The Krebs cycle enzyme aconitase, for example, contains an iron-sulfur (Fe/S) cluster. As discussed later in this chapter, the iron content of the Fe/S cluster of a related aconitase-like molecule allows it to “sense” iron concentrations within the cell and to act as an iron regulatory protein (IRP) to modulate the translation of genes of iron metabolism.
Iron Toxicity
The ability of iron to catalyze redox reactions also accounts for its toxicity. As an enzymatic cofactor, the metal is involved in the restructuring of cellular components. Unchelated iron has unbridled redox activity, however, and may contribute to cellular degeneration. We live in an oxygen-rich atmosphere, and our bodies require oxygen for many metabolic processes. However, oxygen is highly reactive and its interaction with iron potentiates its toxicity. Normal cellular reactions, including respiration, generate the reactive oxygen intermediates superoxide (O 2 − ) and hydrogen peroxide (H 2 O 2 ). Oxidative stress develops when production of reactive oxygen species exceeds the body’s processing capacity. Under these circumstances, reactive oxygen intermediates may be converted to injurious free radicals by the iron-catalyzed Fenton reaction : Hydroxyl radicals promote the peroxidation of proteins, DNA, and membrane lipids, problems that are exacerbated by iron overload. Certain organelles are particularly susceptible to iron-dependent peroxidation. In iron-overloaded cells, injured mitochondria and lysosomes become leaky. Mitochondrial damage and release of lysosomal proteases cause further cell injury and may ultimately lead to cell death. This process contributes to the severe tissue damage seen in the liver, heart, and endocrine organs of patients with iron overload disorders (see later).
Iron is not normally present in cell membranes. However, in both sickle cell disease and thalassemia, iron, heme, ferritin, and denatured hemoglobin adhere to the inner surface of the red cell plasma membrane and thereby contribute to the pathogenesis of these congenital anemias. The membrane complex containing denatured hemoglobin has been termed hemichrome . The red cell anion transport protein band 3 appears to nucleate the formation of these iron aggregates. Injured cells decorated with membrane iron deposits are removed by a functioning spleen in patients with thalassemia and sickle cell–hemoglobin C disease (hemoglobin SC disease), but they circulate in functionally asplenic patients with homozygous sickle cell–hemoglobin S disease (hemoglobin SS disease). Membrane-associated iron promotes free radical formation and further membrane damage, marked by generation of the lipid peroxidation product malonyldialdehyde and by cross-linking of membrane proteins. Membranes become rigid and thus contribute to the formation of irreversibly sickled cells that occlude the microcirculation.
Sometimes reactive oxygen intermediates can be beneficial. Neutrophils contain a membrane-associated reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase that produces superoxide to kill ingested microorganisms (reviewed by Clark ). Superoxide and secondary reactive oxygen intermediates are potent antimicrobial agents. Congenital defects in this NADPH oxidase, collectively termed chronic granulomatous disease (see Chapter 22 ), are characterized by a serious defect in defense against bacterial pathogens.
Neutrophils and iron also injure tissues in inflammatory diseases such as rheumatoid arthritis. Synovial macrophages ingest red cell hemoglobin introduced by intermittent joint hemorrhage. Iron is deposited in the synovial membrane, proximate to superoxide and hydrogen peroxide generated by neutrophils and macrophages participating in the inflammatory reaction. Iron catalyzes the conversion of these compounds to free radical species, which promote lipid peroxidation. Iron therapy exacerbates this process. In contrast, antioxidants and iron chelators retard free radical generation, thereby affording some theoretic protection against injury in rheumatoid arthritis.
These deleterious properties of iron are threatening only when the element is in a “free” state or in an abnormal form within the cell. Protection of cell structures from iron-dependent free radical damage is therefore of utmost importance. When iron is bound to protein either directly or indirectly, the generation of free radicals is largely abrogated. Thus, tight chelation of iron is a means of controlling its reactivity. As discussed later, cytoplasmic ferritin allows iron to be stored safely within cells by sequestering it in an innocuous form. Expression of ferritin is induced by oxidative stress. Both prokaryotic and eukaryotic cells contain ferritins, and mice lacking one of the two ferritin genes do not develop past the early embryonic stages. Thus, ferritin appears to be necessary for most, if not all, living cells.
Acquisition and Distribution of Iron
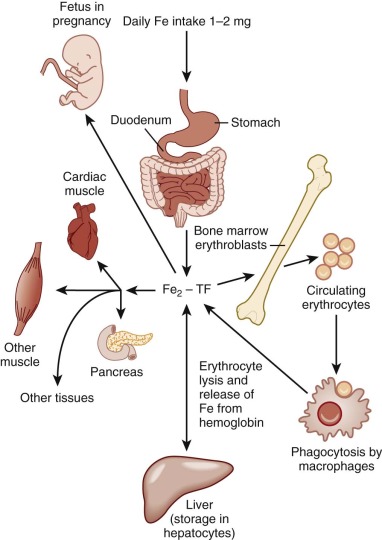
The average adult has 4 to 5 g of iron. To maintain systemic iron homeostasis, a meticulous balance exists between dietary uptake and loss. Approximately 0.5 to 1 mg of iron is lost each day through sloughing of cells from skin and mucosal surfaces ( Fig. 11-1 ). Because menstruating females lose an average of an additional 1 mg of iron daily, their dietary iron requirement is increased. Neither the liver nor the kidney has a significant physiologic capability to excrete iron. Consequently, absorption is the primary means of regulating body iron stores. During neonatal and childhood growth spurts, iron requirements increase in response to augmentation of body, and particularly red blood cell, mass.
Iron Absorption
Iron absorption occurs predominantly in the proximal duodenum. The physical state of iron entering the duodenum greatly influences its absorption. At neutral pH, ferrous iron is rapidly converted to the insoluble ferric form. Acid produced by the stomach serves to lower the pH in the duodenum and thereby enhance the solubility and uptake of iron (see later). Heme is absorbed separately from and more efficiently than inorganic iron, independent of duodenal pH. Consequently, meat is an excellent nutritive source of iron. Heme iron absorption is, however, very poorly understood, but a heme oxygenase inhibitor has been shown to block heme catabolism in the intestine and result in an iron-deficient state.
A number of dietary factors influence inorganic iron absorption. Ascorbate and citrate increase iron uptake, in part by acting as weak chelators to help solubilize the metal in the duodenum. Iron is readily transferred from these compounds to the absorptive epithelium. Conversely, plant phytates, bran, and tannins inhibit iron absorption. These compounds chelate iron and prevent its uptake by the absorption machinery.
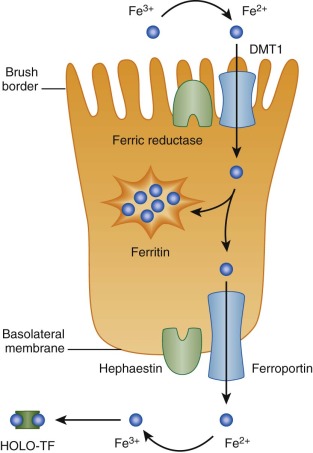
Through a combination of genetic and biochemical approaches, much has been learned about the absorption of nonheme iron over the past two decades. Nonheme iron arrives at the apical surface of the absorptive duodenal enterocyte in its ferric (Fe 3+ ) form. It is reduced through the action of a brush border ferric reductase. This enzyme may be duodenal cytochrome b , a heme protein that is homologous to b 561 cytochromes. Expression of duodenal cytochrome b is significantly greater in the proximal duodenum than elsewhere and increases in iron deficiency.
Ferrous (Fe 2+ ) iron is then taken up by the enterocyte through the action of divalent metal transporter 1 (DMT1, also known as Nramp2, DCT1, and SLC11A2). Transport requires the coordinated movement of protons in the same direction (symport). Thus, DMT1 functions only at low pH, and it has little or no activity at neutral pH. DMT1 is widely expressed, but duodenal levels increase dramatically in iron deficiency.
DMT1 can also transport other divalent metal ions, including Cd 2+ , Co 2+ , Cu 2+ , Mn 2+ , Pb 2+ , and Zn 2+ . Competition studies have shown that lead, manganese, cobalt, and zinc can share the intestinal absorption pathway used by iron. Increased iron absorption induced by iron deficiency also enhances the uptake of these elements. Because iron deficiency often coexists with lead intoxication, this interaction has vast public health significance and can produce particularly serious medical complications in children. As discussed later, copper absorption and metabolism are handled by an entirely different mechanism.
After iron enters the absorptive enterocyte through the action of DMT1, it has at least two possible fates. It can be retained by the cell and subsequently be lost when the enterocyte dies and is sloughed into the intestinal lumen, or it can be transported across the basolateral membrane to enter the body. Iron retained by the enterocyte is used for cellular metabolism or incorporated into ferritin. Exported iron leaves the cell by way of a unique basolateral transmembrane iron transporter, ferroportin (FPN1, also known as SLC40A1, MTP1, and IREG1).
Basolateral iron transfer also requires a change in the oxidation state of the metal, likely facilitated by the homologous multicopper ferroxidases hephaestin, and ceruloplasmin, which are membrane associated and present in the plasma, respectively. A comprehensive model of intestinal iron absorption, primarily derived from studies in mice, is shown in Figure 11-2 . This model pertains only to nonheme iron transport; details of heme iron uptake have not yet been worked out. A candidate intestinal heme transporter was reported but subsequently proven to be a folate transporter, instead.
Normally, only about 10% of dietary nonheme iron entering the duodenum is absorbed. However, this value increases significantly with iron deficiency. In contrast, iron overload reduces, but does not completely eliminate, absorption. Finch designated this modulation the “stores regulator.” In addition, both iron deficiency anemia and forms of anemia associated with ineffective erythropoiesis induce a marked increase in iron absorption. This effect has been designated the “erythroid regulator.” The erythroid regulator appears to exert a dominant effect over the stores regulator, as individuals with chronic persistent ineffective erythropoiesis can become profoundly iron overloaded even in the absence of transfusion. Hypoxia also increases iron absorption, independent of anemia.
Hepcidin, an iron-regulated peptide hormone, links the actions of all these regulators through a unifying molecular mechanism. Hepcidin is a 25–amino acid peptide produced in the liver from a larger precursor (reviewed by Nemeth and Ganz ). It circulates in the plasma and binds to the iron exporter ferroportin on the basolateral surface of absorptive enterocytes, causing ferroportin to be internalized and degraded in a ubiquitin-dependent manner. In this way, hepcidin controls cellular iron egress. Hepcidin thus regulates iron absorption at the level of the intestinal epithelium in that any iron unable to leave the enterocytes is lost when these cells turn over.
Expression of hepcidin is induced in response to iron overload and inflammation and is repressed in response to increased erythropoietic activity and hypoxia. Regulation of hepcidin levels appears to be at the level of gene transcription. However, the signals that govern hepcidin transcription in response to these stimuli are incompletely understood. Only the inflammatory cytokine interleukin-6 has been definitely shown to be involved in regulation of hepcidin expression by physiologic changes outside the liver.
Most of the total body iron is ultimately incorporated into hemoglobin in erythroid precursors. An average adult produces 200 billion red cells daily to achieve a red cell renewal rate of 0.8% per day. Each red cell contains more than 1 billion atoms of iron, and each milliliter of packed red blood cells contains approximately 1 mg of iron. To meet this daily need for 2 × 10 20 atoms (or 20 mg) of elemental iron, iron is recycled from senescent red cells and returned to the circulation. Plasma iron turnover (PIT) represents the mass turnover of transferrin-bound iron in the circulation (expressed as milligrams per kilogram per day). Accelerated erythropoiesis increases PIT and enhances iron uptake from the gastrointestinal tract. Although hepcidin appears to be the effector molecule that alters intestinal absorption, an upstream, circulating factor—the erythroid regulator—that communicates the iron needs of the erythron must exist. There are some data to indicate that circulating bone morphogenetic protein (BMP) signaling pathway inhibitors (e.g., GDF15 and TWSG) may play a role in mediating this effect.
This erythroid control of hepcidin expression is particularly apparent in patients with thalassemia intermedia (non–transfusion-dependent thalassemia), in whom marked iron overload develops even without transfusions. The accelerated, but ineffective, erythropoiesis substantially boosts iron absorption by inhibiting hepcidin expression. By contrast, hepcidin is increased in patients with thalassemia and other anemias receiving chronic transfusion therapy as a consequence of iron overload and the suppression of erythropoiesis.
Intercellular Iron Transport
Only a small proportion of total body iron enters and leaves the body each day. Consequently, intercellular iron transport is quantitatively more important than intestinal absorption. The greatest mass of iron is found in erythroid cells, which make up about 60% to 80% of the total body endowment in normal individuals. To accommodate ongoing erythropoietic needs, the reticuloendothelial system recycles approximately 20 to 25 mg of iron from effete red cells each day.
A small fraction, 0.1% or 4 mg, of total body iron circulates in plasma in an exchangeable pool. In normal individuals, nearly all of the circulating plasma iron is bound to transferrin. Transferrin serves three purposes: (1) it renders iron soluble under physiologic conditions; (2) it prevents iron-mediated free radical toxicity; and (3) it facilitates transport into cells. Transferrin is by far the most important physiologic supplier of iron to red cells. In fact, plasma transferrin serves to deliver iron to most tissues of the body. It is an 80-kd glycoprotein that has homologous N-terminal and C-terminal iron-binding domains. The molecule is related to several other proteins, including ovotransferrin in bird and reptile eggs, lactoferrin in extracellular secretions and neutrophil granules, and melanotransferrin (p97), a protein produced by melanoma cells. The functions of these related proteins are incompletely understood. The human transferrin messenger RNA (mRNA) is 2.3 kilobases (kb) in length and encodes 679 amino acids, including a 19–amino acid leader sequence. It is located on chromosome 3q, near genes for the transferrin receptor and melanotransferrin. The liver is the major site of synthesis and secretion of transferrin. Other tissues, including Sertoli cells of the testis, oligodendrocytes of the central nervous system, lymphocytes, muscle cells, and mammary cells, can, however, also produce the protein. Local synthesis within the brain and testis apparently provides transferrin for these tissues because serum transferrin does not penetrate their unique capillary barriers.
Transferrin production is regulated at multiple levels. Several cis -acting control regions exist upstream of the gene. The transferrin promoter contains binding sites for tissue-specific nuclear factors that activate transcription differentially in the liver and other tissues (e.g., Sertoli cells). Iron, hormones, and inflammatory stimuli also modulate transferrin gene expression. In the setting of iron deficiency, serum transferrin levels rise substantially as a result of enhanced synthesis of transferrin mRNA by the liver. In contrast, inflammation depresses levels of both serum transferrin and serum iron, the latter through the action of the iron regulatory hormone hepcidin. The plasma concentration of diferric transferrin has been proposed to modulate expression of the iron regulatory hormone hepcidin. Liver-derived serum transferrin levels fall modestly in patients with genetic or acquired iron overload, although mRNA levels have been reported to be unchanged. This suggests that hepatic transferrin is also controlled at the level of translation or secretion. The quantity of transferrin mRNA in nonhepatic tissues (including the testis, kidney, and spleen) is not affected by iron deficiency. The liver-derived transferrin, therefore, appears to have the unique responsibility of responding to iron status.
X-ray crystal structures have been determined for transferrin and related proteins (reviewed elsewhere ). All members of the transferrin protein superfamily exhibit similar polypeptide folding. The N-terminal and C-terminal domains are globular moieties of about 330 amino acids; each of these domains is divided into two subdomains, with the iron- and anion-binding sites located in the intersubdomain cleft on either side of a central plane of symmetry, suggesting an origin by gene duplication from a primordial protein containing a single iron-binding site. The binding cleft opens with iron release and closes with iron binding. The N-terminal and C-terminal binding sites are very similar. No cooperativity exists in binding of iron by the two sites, and the protein can be proteolytically cleaved into two halves, each of which retains iron-binding capability. Transferrin binds ferric iron much more avidly than ferrous iron.
The precise mechanism by which iron is loaded onto transferrin as it leaves intestinal epithelial cells or reticuloendothelial cells is unknown. The copper-dependent ferroxidase ceruloplasmin and its homologue hephaestin likely play central roles in reducing ferrous iron exported by ferroportin 1 (FPN1) from the reticuloendothelial cells and intestine, respectively, although neither is absolutely required due to the tendency of iron to oxidize in aqueous environments. Transferrin binds ferric iron exceptionally avidly, with a dissociation constant ( K d ) of approximately 10 to 22 mol/L in the company of an anion (usually carbonate), which serves as a bridging ligand between the metal and protein and excludes water from two coordination sites. Without the anion cofactor, iron binding is negligible; with it, ferric transferrin is resistant to all but the most potent chelators. Release of iron from transferrin involves protonation of the carbonate anion to loosen the metal-protein association.
The sum of all iron-binding sites on transferrin constitutes the total iron-binding capacity (TIBC) of plasma. Thus, on a molar basis, TIBC is twice the concentration of transferrin protein because each transferrin molecule can bind two iron atoms. The laboratory method used to measure the TIBC can either be truly the total iron-binding concentration of the plasma measured by iron binding or a calculation based upon measurement of the concentration of transferrin. Whatever the method, under normal circumstances, about a third of transferrin iron-binding sites are filled. Consequently, except for the situation of extreme iron overload, in which all transferrin-binding sites are occupied, non–transferrin-bound iron (NTBI) in the circulation is present at very low concentrations. The distribution of plasma and tissue iron can be traced with the use of 59 Fe as a radioactive tag by reinfusing a subject with autologous transferrin loaded with radiolabeled iron. Blood samples can be analyzed at timed intervals to determine the rate of loss of the radioactive label. Such ferrokinetic studies indicate that the normal half-life of iron in the circulation is about 75 minutes. The absolute amount of iron released from transferrin per unit time is the PIT (see earlier).
Such radioactive tracer studies indicate that at least 80% of the iron bound to circulating transferrin is delivered to the bone marrow and incorporated into reticulocytes. Other major sites of iron delivery include the liver and the spleen, which are the primary depots for iron storage. Hepatic iron is found in both the reticuloendothelial cells and hepatocytes. Reticuloendothelial cells acquire iron primarily by phagocytosis and breakdown of effete red blood cells; they catabolize heme and return iron to the circulation, where it binds transferrin. Hepatocytes take up iron by at least two different mechanisms, which are transferrin-dependent and transferrin-independent. The relative amounts of iron in each of these cell types depend on clinical circumstances, as discussed in detail later.
Given the preeminent role of bone marrow in the clearance of labeled iron from the circulation, ferrokinetic studies provide a window on erythropoietic activity. Conditions that augment erythrocyte production increase PIT. For example, hemolytic anemias such as hereditary spherocytosis and sickle cell disease induce rapid delivery of transferrin-bound iron to the marrow. In contrast, disorders that reduce red cell production, such as Diamond-Blackfan anemia and aplastic anemia, prolong PIT.
When erythrocytes are produced and released into the circulation in a normal fashion, the process of erythropoiesis is termed effective . In patients with certain anemias, however, the abnormal, nascent red cells apoptose or are destroyed before they leave the marrow cavity. In this situation, erythropoiesis is ineffective , which means simply that the erythropoietic precursors have failed to accomplish their primary task: delivery of mature, enucleated erythrocytes to the circulation. The ferrokinetic profile in this case shows rapid removal of iron from transferrin with delayed entry of label into the pool of circulating red cell hemoglobin. β + -thalassemia is an important example of this pattern. In β + -thalassemia, ineffective erythropoiesis is associated with a markedly enhanced PIT.
Intracellular Iron Metabolism
Transferrin and the Transferrin Cycle
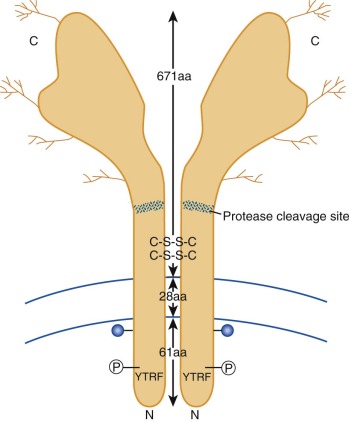
Although transferrin was first characterized more than 60 years ago, its receptor eluded investigators until the early 1980s, when monoclonal antibodies prepared against tumor cells were found to recognize the transferrin receptor glycoprotein. Subsequently, receptor-mediated endocytosis of iron bound to transferrin has been characterized in detail. A diagram showing key features of the transferrin receptor is presented in Figure 11-3 . Each subunit of the disulfide-linked homodimer contains 760 amino acids. Oligosaccharides account for about 5% of the 90-kd subunit’s molecular mass. Four glycosylation sites (three N linked and one O linked) are found in the protein. Glycosylation-defective mutants have fewer disulfide bridges, less transferrin binding, and less cell surface expression. The transmembrane domain, located between amino acids 62 and 89, functions as an internal signal peptide because there is none at the N-terminal end. A molecule of fatty acid (usually palmitate) is also covalently linked to each subunit at the internal edge of the transmembrane domain and may play a role in membrane localization. The crystal structure of the ectodomain of the transferrin receptor has been reported. Although no one has yet succeeded in cocrystallizing the receptor with transferrin, molecular modeling and binding studies of mutant transferrin receptor proteins have yielded important insights, including the amino acid residues important for receptor binding to transferrin.
Iron is taken into cells by receptor-mediated endocytosis of diferric transferrin ( Fig. 11-4 ). Receptors on the outer face of the plasma membrane bind iron-loaded transferrin with very high affinity. The C-terminal domain of transferrin appears to mediate receptor binding. Diferric transferrin binds with much higher affinity than monoferric transferrin or apotransferrin does. The dissociation constant ( K d ) for bound diferric transferrin ranges from 10-7 to 10-9 mol/L at physiologic pH, depending on the species and tissue. The K d of monoferric transferrin is approximately 10-6 mol/L. The concentration of circulating transferrin is about 25 µmol/L. Therefore, cellular transferrin receptors are ordinarily fully saturated.
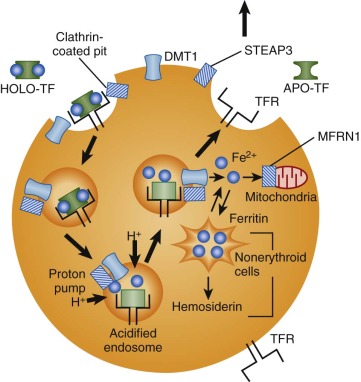
After binding to its receptor on the cell surface, transferrin is internalized through a constitutive mechanism that begins with invagination of clathrin-coated pits and the formation of endocytic vesicles. This process requires the short, 61–amino acid intracellular tail of the transferrin receptor molecule. Receptors with truncated N-terminal cytoplasmic domains do not recycle properly. This portion of the molecule contains a conserved tyrosine-threonine-arginine-phenylalanine (YTRF) sequence that functions as a signal for endocytosis.
An unknown ATP-dependent proton pump lowers the pH of the internalized endosome to about 5.5. Acidification of the endosome weakens the association between iron and transferrin and promotes a conformational change in the transferrin receptor to enhance binding of apotransferrin and facilitate release of iron. An endosomal ferrireductase, STEAP3, reduces iron from the Fe 3+ state to Fe 2+ , either at the same time that it is released from transferrin or soon afterward. STEAP3 is a membrane-bound NADPH-dependent oxidoreductase localized to transferrin cycle endosomes and expressed at high levels in erythroid precursors. Related proteins may serve similar functions at other sites.
The iron released from transferrin must exit the endosome and enter the cytoplasm and mitochondria for use in heme biosynthesis, Fe/S cluster biosynthesis, and storage and for other purposes. This transmembrane transport step is also mediated by DMT1. DMT1 is unusual in that it is expressed on two very different types of membranes—the apical membrane of intestinal enterocytes and the endosomal membrane of transferrin uptake vesicles in nonpolarized cells. Once transported out of the endosome or across the plasma membrane, iron must be delivered to sites of use or stored in the form of ferritin. The cytosolic chaperone that traffics iron to ferritin appears to be PCBP1, a protein that also functions in the control of mRNA stability and translation, particularly in erythroid cells. In erythroid cells, iron is delivered across the mitochondrial inner membrane by mitoferrin 1 (MFRN1 or SLC25A37) ; once across the membrane it is incorporated into heme by the enzyme ferrochelatase.
The fate of transferrin and the transferrin receptor is distinct from that of iron. Rather than entering lysosomes for degradation, as do the ligands in other receptor-mediated endocytosis pathways, intact receptor-bound apotransferrin recycles to the cell surface, where the neutral pH promotes detachment into the plasma. Thus, preservation and reuse of transferrin are accomplished by pH-dependent changes in the affinity of transferrin for iron and the transferrin receptor. Exported apotransferrin binds additional iron atoms and undergoes further rounds of iron delivery to cells. Thus, a single transferrin molecule, with a half-life of 8 days, may be used hundreds of times for delivery of iron.
It was once a widely accepted belief that the transferrin cycle was important, if not necessary, for the uptake of iron by all mammalian cells. However, two lines of evidence indicate that such is not the case. First, mice and humans with severe deficiencies of plasma transferrin have iron deficiency anemia but vastly increased amounts of iron in nonhematopoietic tissues. This finding indicates that uptake of iron bound to transferrin is important for red blood cell hemoglobinization and maturation but that transferrin is not essential for delivery of iron to most other tissues. Second, mouse embryos lacking transferrin receptor die midway through gestation as a result of severe anemia, but most nonhematopoietic tissues appear generally intact. Aside from the erythron, only the developing central nervous system shows an apparent requirement for transferrin receptor. In its absence, primitive neuroepithelial cells undergo apoptosis at a greatly increased rate. Taken together, these observations indicate that the transferrin cycle is of primary importance in erythropoiesis and neurogenesis but of lesser importance in other mammalian tissues, at least early in embryonic development.
Non–Transferrin-Bound Iron Uptake
Both hypotransferrinemia and iron overload lead to complete saturation of available plasma transferrin, and non–transferrin-bound iron (NTBI) circulates in a chelatable, low–molecular-weight form (reviewed by Cabantchik and colleagues ). This iron is weakly complexed to albumin, citrate, amino acids, sugars, and other small molecules, and it behaves differently from iron associated with transferrin. Nonhematopoietic tissues (particularly the liver, endocrine organs, kidneys, and heart) preferentially take up NTBI. Radiolabeled iron administered to mice with and without available transferrin-binding capacity is distributed in markedly different patterns. In normal animals with excess plasma iron-binding capacity, hematopoietic tissues are the primary sites of uptake. When free transferrin iron-binding sites are absent, however, most iron is deposited in the liver and pancreas, thus indicating that these organs serve as the initial iron reservoirs in the situation of iron overload. Notably, this pattern of distribution is similar to that seen in idiopathic hemochromatosis. NTBI is highly toxic to cells. Plasma NTBI can potentiate the formation of free radicals through the Fenton reaction (see earlier) and thereby induce cell membrane damage. Cardiac cells are particularly susceptible to this damage. Hence, therapeutic chelation must effectively remove plasma NTBI.
The identity or identities of the protein(s) responsible for NTBI uptake are of considerable interest due to the clinical impact of NTBI uptake in hemochromatosis, iron-loading anemias, and chronic transfusion therapy. NTBI uptake activities have been characterized in tissue culture cells, but the physiologically relevant transporters are only beginning to be understood. Topologically, the cell exterior and the endosome interior are equivalent compartments. The primary role of the transferrin–transferrin receptor interaction is to sequester iron near the cell membrane, thereby increasing the likelihood of iron uptake. DMT1 may reside on the plasma membrane of the cell before endocytosis. If so, it should be oriented to transport iron directly into the cell, without the assistance of transferrin (as diagrammed in Fig. 11-4 ). However, the fact that DMT1 functions only at acidic pH suggests that it probably does not, by itself, account for all NTBI transport. Indeed, hepatocyte DMT1 expression was recently shown not to be essential for hepatocellular iron accumulation in the setting of iron overload. There is some evolving in vivo evidence to suggest that L-type calcium channels, or the zinc transporter family (ZIP) proteins ZIP8 and ZIP14 can transport NTBI.
Role of Iron in Cell Proliferation and Differentiation
Iron is indispensable for DNA synthesis and a host of metabolic processes. Iron starvation arrests cell proliferation, presumably because ribonucleotide reductase and other enzymes require the metal. Although transferrin receptors are expressed on all dividing cells in numbers that roughly reflect their growth rate, as discussed in detail earlier, the erythron is the tissue that relies most heavily on delivery of iron by transferrin,. However, the transferrin cycle also appears to play a significant role in other cell types.
Studies of mature T lymphocytes exemplify the general relationship between expression of transferrin receptor and cell proliferation. Transferrin receptors, absent from resting T cells, have long been recognized as markers of T-cell activation. Initiation of cell division by a mitogen such as phytohemagglutinin dramatically increases surface expression of both transferrin receptor and iron uptake. Furthermore, tumor cells upregulate transferrin receptor expression to optimize acquisition of iron for proliferation, and certain monoclonal antibodies against the transferrin receptor curb proliferation of tumor cells in vitro and in vivo. Interestingly, some reports have suggested that the transferrin receptor may have an additional role in activated T cells, apart from its iron uptake function. Anti–transferrin receptor monoclonal antibodies have been described that can trigger T-cell activation and secretion of interleukin-2. These antibodies presumably activate a signal transduction pathway beginning with the transferrin receptor but independent of iron trafficking. The transferrin receptor also appears to have a role in early lymphocyte development. Embryonic stem cells in which both transferrin receptor genes have been inactivated fail to differentiate into circulating lymphocytes in vivo in chimeric mice. It is not yet clear whether this is due to defective iron delivery or to perturbation of some other, as yet unknown function of the transferrin receptor.
Chelators that deplete intracellular iron and limit its bioavailability in extracellular fluids further demonstrate the central role of iron in cell proliferation. Agents such as deferoxamine, desferrithiocin, and pyridoxal isonicotinoyl hydrazone inhibit the growth of a variety of tumor cells in culture and greatly reduce T-cell proliferation. A likely inhibitory mechanism is iron deprivation, with reduced ribonucleotide reductase activity and lower levels of deoxyribonucleotides. This, in turn, leads to mitotic arrest in the S phase of the cell cycle. The addition of iron to the medium reverses the growth inhibition. Chelators may also induce apoptosis, or programmed cell death, through other mechanisms that are not yet understood.
Above all cells, erythroid precursors need an extraordinary amount of iron to support hemoglobin synthesis and differentiation into mature red cells. The density of transferrin receptors on the cell surface is modulated during erythroid development. Transferrin receptors first appear in measurable numbers on colony-forming unit–erythroid (CFU-E) cells and increase to 300,000 per cell on proerythroblasts and as many as 800,000 per cell on basophilic erythroblasts at the time of maximal iron uptake. Numbers then fall to 100,000 per cell on circulating reticulocytes and to negligible levels on mature red cells. A precise correlation exists between iron requirement and transferrin receptor number, thus indicating that the abundance of transferrin receptors on the cell surface is a major determinant of erythroid iron uptake. In culture, a monoclonal antibody to the transferrin receptor that permits ligand binding but subsequently slows receptor recycling partially blocks erythroid iron uptake. The level of iron uptake is sufficient for cell division but not hemoglobin synthesis.
A wealth of literature demonstrates that oxidized heme (hemin) promotes differentiation of erythroleukemia cell lines in tissue culture. Conversely, deficient heme biosynthesis abrogates chemical induction of differentiation in an erythroleukemia cell line subclone. Heme regulates the transcription of globin, transferrin receptor, and ferritin genes. Some of these effects may be mediated by the heme-dependent transcriptional repressor BACH1. A second, major mechanism in which heme regulates terminal erythroid differentiation is mediated by a heme-dependent elongation factor 2a kinase termed heme-regulated kinase (HRI), which globally suppresses translation in erythroid cells in the absence of heme (reviewed by Chen ). Although some of the precise mechanisms remain to be elucidated, it is clear that iron uptake, heme biosynthesis, and globin protein production are coordinately regulated. Interrelated regulatory networks apparently allow red cell precursors to maximize hemoglobin formation without accumulating excess globin proteins, unbound iron, or toxic protoporphyrin intermediates.
Regulation of Proteins of Iron Metabolism
Ferritin
Once inside the cytoplasm, iron is almost certainly bound by carrier molecules that assist in delivering iron to intracellular locations, including mitochondria (for heme biosynthesis) and ferritin (for storage). The identities of the intracellular iron carrier molecules by and large remain unknown. However, there is recent evidence to suggest that poly-C–binding protein 1 (PCBP1), which is involved in mRNA stability and translation, also serves as an iron chaperone specific for iron acquisition by ferritin and the hypoxia-inducible factor (HIF) prolyl hydroxylase. The amount of iron in transit within the cell at any given time is small and difficult to measure. This minute pool of transit iron, which is believed to be in the Fe 2+ oxidation state, is the biologically active and potentially toxic form of the element. Metabolically inactive iron, stored in ferritin and hemosiderin, is nontoxic and in equilibrium with this exchangeable transit iron.
Both prokaryotes and eukaryotes produce ferritin molecules for iron storage. Mammalian ferritin molecules are complex 24-subunit heteropolymers of H (for heavy or heart) and L (for light or liver) protein subunits. L subunits are 19.7 kd in mass, with isoelectric points of 4.5 to 5; H subunits are 21 kd in mass, with isoelectric points of 5 to 5.7. The subunits of the ferritin molecule probably arose by gene duplication, although the degree of nucleotide sequence homology between the two is only about 50%. They assemble to form a sphere with a central cavity in which up to several thousand atoms of crystalline iron can be stored in the form of poly-iron-phosphate oxide. Eight channels through the sphere are lined by hydrophilic amino acid residues, and six more are lined by hydrophobic residues. Strong interspecies amino acid conservation is seen in the residues that line the hydrophilic channels, whereas marked variation is seen in those along the hydrophobic passages. The hydrophilic channels terminate with aspartic acid and glutamic acid residues and are lined by serine, histidine, and cysteine residues, all of which potentially bind metal ligands.
Although the two ferritin chains are homologous, only H ferritin has ferroxidase activity, which is essential for iron incorporation into the ferritin polymer. Isoelectric focusing shows that ferritin polymers have a variable composition of H and L subunits. These isoferritins, as they are called, show tissue-specific variation. Ferritin from liver, for instance, is rich in L subunits, as is that from the spleen. In contrast, the heart has ferritin rich in H subunits. Increased H-subunit content correlates with increased iron use, whereas increased L-subunit content correlates with increased iron storage. The ratio of H to L subunits increases with increased cell proliferation. Thus, ferritin provides a flexible reserve of iron.
Despite the fact that it does not, itself, have enzymatic activity, a mutation in L ferritin has been shown to cause a distinctive iron deposition disease in humans termed neuroferritinopathy . In patients carrying a nucleotide insertion altering the carboxy-terminus of L ferritin, iron overload develops in the basal ganglia and leads to a neurodegenerative disease with variable extrapyramidal features. By contrast, pathologic accumulation of apo-L-ferritin leads to a distinctive form of hereditary cataracts (hereditary hyperferritinemia-cataract syndrome) that is unassociated with iron metabolism abnormalities (see below). A number of other disorders, including aceruloplasminemia (see below), as well as pantothenate kinase deficiency, can also lead to pathologic iron deposition in distinctive regions of the brain.
Ferritin molecules aggregate into clusters that are engulfed by lysosomes and degraded. The end product of this process, hemosiderin, is an amorphous agglomerate of partially denatured protein and lipid interspersed with iron oxide molecules. In cells overloaded with iron, lysosomes accumulate large amounts of hemosiderin, which can be visualized by Prussian blue (Perls) staining. Although the iron enmeshed in this insoluble compound constitutes an end-stage product of cellular iron storage, it remains in equilibrium with cytosolic ferritin. Ferritin iron, in turn, is in equilibrium with iron complexed to low–molecular-weight carrier molecules. Therefore, introduction of an effective chelator into the cell captures iron from the low–molecular-weight “toxic” iron pool, draws iron out of ferritin, and eventually depletes iron from hemosiderin as well, though only very slowly. As might be expected, the bioavailability of hemosiderin iron is much lower than that of iron stored in ferritin.
The large number of processed pseudogenes that exist for each subunit initially confounded pinpointing the chromosomal location of ferritin genes. Functional ferritin genes for the H and L subunits are located on human chromosomes 11 and 19, respectively. In addition, there is an intronless gene encoding a mitochondrial, H-like ferritin molecule located on human chromosome 5q. The function of mitochondrial ferritin remains uncertain, although the fact that its expression is increased in erythroid precursors from patients with sideroblastic anemia suggests that it is involved in mitochondrial iron management.
Ferritin formation is controlled at multiple levels—transcription, message stabilization, translation, and subunit assembly. In the liver and in HeLa cells, iron rapidly induces the synthesis of L-subunit mRNA, with no effect on H-subunit transcription. In contrast, induced differentiation of HL-60 promyelocytic leukemia cells and mouse erythroleukemia cells increases production of H-subunit mRNA. Tumor necrosis factor induces H-chain transcription in human myoblasts. Iron, heme, reactive oxygen species, and oxidative stress all enhance transcription and translation of the ferritin heavy chain (reviewed by Rouault ).
Cytoplasmic ferritin mRNA forms a stable complex with several proteins. Both iron and interleukin-1β enhance translation of ferritin mRNA. Influx of iron into cells shifts the message onto the ribosomes, thereby enhancing the synthesis of ferritin subunits. This translational control mechanism involves an RNA-protein interaction that links the expression of genes encoding ferritin, the transferrin receptor, enzymes of heme biosynthesis, DMT1, ferroportin, and other proteins. Zahringer, Baliga, and Munro initially showed that ferritin synthesis was regulated at the level of message translation. Comparison of the 5′untranslated regions of ferritin mRNA encoding both heavy and light chains showed striking conservation of a 28-bp sequence motif that was predicted to form a stable RNA stem-loop structure and was necessary for translational control of ferritin ( Fig. 11-5 , inset).
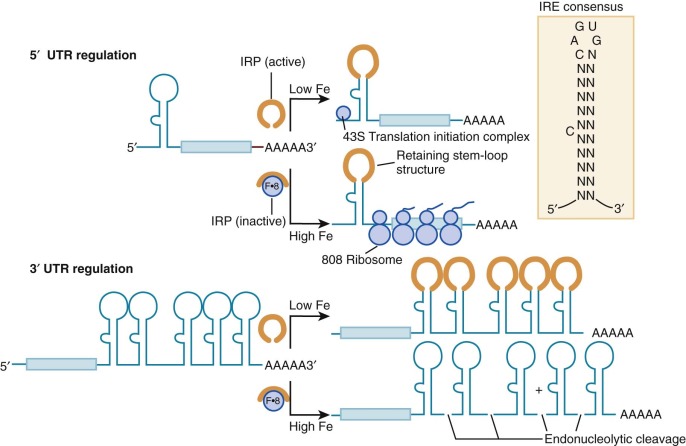
Subsequently designated the iron response element (IRE), this RNA stem-loop is recognized by at least two specific RNA-binding proteins called IRP1 and IRP2 for iron regulatory proteins 1 and 2. IRP1 is a 98-kd soluble polypeptide with homology to the mitochondrial tricarboxylic acid–cycle enzyme aconitase (ACO1). Indeed, IRP1 is the cytoplasmic isozyme of aconitase. Like mitochondrial aconitase, the enzymatic active site of IRP1 contains a 4Fe/4S cluster. When the iron concentration in the cytosol is high, the cluster is complete and the protein is in its holoenzyme form and possesses aconitase activity and cannot bind mRNA. Under these conditions, the ferritin message is translated efficiently. Conversely, when the iron concentration is low, the protein is in its apo-form and aconitase activity is absent, RNA binding is avid, and ferritin message translation is blocked because translation initiation complexes cannot form properly. Thus, IRP1 is a dual-function protein, with ambient iron controlling the switch by assembly and disassembly of a 4Fe/4S cluster. Other molecules that modulate the switch include ascorbic acid, nitric oxide, and heme, all of which contribute to the oxidation status of the cell. This mechanism allows ferritin protein expression to be rapidly downregulated without degradation of mRNA. The importance of IRP-mediated translational control is underscored by the fact that plasma hyperferritinemia and ferritin-containing cataracts develop in patients with mutations in the L-ferritin IRE. Nevertheless, these patients with hereditary hyperferritinemia cataract syndrome do not have abnormalities in iron homeostasis. Furthermore, it appears that an iron overload disorder resembling hereditary hemochromatosis develops in rare patients with mutations in the H-ferritin IRE, presumably because increased H-ferritin expression leads to increased cellular iron assimilation or retention, or both.
A second IRE-binding protein, IRP2, also modulates posttranscriptional expression of mRNA by binding to IREs in much the same way. Initially isolated in the liver, IRP2 has been found in all tissues examined and is more abundant than IRP1 in some. IRP2 lacks aconitase activity and does not contain an Fe/S cluster to function as an iron sensor. Rather, IRP2 protein levels are modulated directly by iron or by heme and the molecule is actively degraded when iron or heme (or both) is abundant. Recently, it has been shown that an E3 ubiquitin ligase complex that contains FBXL5 controls IRP2 protein abundance by targeting it for proteasomal degradation. FBXL5 contains an iron- and oxygen-binding hemerythrin domain that acts as a ligand-dependent regulatory switch that mediates the stability of FBXL5 under iron- and oxygen-replete conditions.
The Transferrin Receptor
IRPs also modulate expression of the transferrin receptor gene. The gene encoding the transferrin receptor is located on human chromosome 3q26.2-qter, near the gene encoding transferrin at 3q21. It consists of 19 exons spread over 31 kb. The transferrin receptor mRNA is approximately twice the length needed to encode the receptor protein. Its lengthy 3′untranslated region contains five RNA stem-loop structures that are structurally similar to those of the ferritin IREs. IRP binding to these sites increases the stability of the transferrin receptor message by obscuring an endonucleolytic cleavage site (see Fig. 11-5 ). The result is a larger amount of transferrin receptor mRNA in the cell. A deficit of cellular iron raises transferrin receptor mRNA levels at least in part through enhanced message stability. However, IRE regulation is probably not responsible for the primary increases in transferrin receptor number in erythroid cells, because this occurs at the transcriptional level and is unaffected by iron status. Relatively little is understood about the tissue-specific transcriptional regulation of the transferrin receptor.
The versatile regulatory functions of IREs may also confer iron-dependent regulation on other proteins. IREs are present in the 5′ untranslated regions of the mRNA encoding the erythroid form of the heme biosynthetic enzyme δ-aminolevulinic acid synthase, mitochondrial aconitase, and ferroportin. A single 3′ IRE is found in one splice isoform of DMT1 mRNA. As the rate-limiting enzyme in heme biosynthesis, the IRE in the erythroid form of δ-aminolevulinic acid synthase produces a conceptually satisfying link between iron and heme production. However, the functions of the IREs in the other mRNAs are not yet understood. It is postulated that all IREs serve the common purpose of coupling changes in the iron status of the cell with its ability to use and store the element (see Fig. 11-5 ). When the intracellular iron concentration is low, the IRE of ferritin mRNA binds IRP and reduces ferritin synthesis because additional iron storage capacity is not needed in this circumstance. Simultaneously, the level of transferrin receptor mRNA increases as the IRP stabilizes the message, thereby increasing expression of the transferrin receptor. Together, these events increase the flow of iron into cells while protecting against iron toxicity. When iron levels in the cell are high, the opposite scenario is operative. Although the IRPs accomplish these regulatory feats under extremes of iron status, the importance of IREs and IRPs in modulating gene expression in normal humans and animals is not fully understood. Recently, novel IRE-containing genes have been identified, with roles less directly related to iron homeostasis.
In an attempt to sort out the unique functions of IRP1 and IRP2 and to evaluate their roles in vivo, targeted mutagenesis was used to generate mutant mice lacking each of the proteins. Initial characterization of IRP1 knockout mice indicated that they had no identifiable defects, but more recent studies have shown that the animals have a transient polycythemia that is associated with an elevated erythropoietin level due to dysregulation of HIF-2α translation, providing further evidence for crosstalk between hypoxia-sensing and iron-regulatory pathways. IRP2 knockout mice have abnormalities in several tissues. Initially, the most striking aspect of the IRP2 knockout phenotype was a late-onset neurologic disorder associated with abnormal deposition of iron in white matter tracts and nuclei throughout the brain. The mice demonstrated ataxia, bradykinesia, and tremors. However, these findings could not readily be explained by known activities of IRP2, and a second group reported that an independently targeted line of mice did not have obvious neurodegeneration. It is possible that the discrepancy may be explained by differential severity of a neurologic phenotype on different genetic backgrounds. More recently, both groups have reported that IRP2-deficient mice have mild microcytosis and erythropoietic protoporphyria as well as subtle perturbations in systemic iron homeostasis.
Serum Ferritin and Soluble Transferrin Receptors
Although most ferritin is located within cells, a measurable amount of the protein exists in serum. Intracellular concentrations, particularly in the liver, are several orders of magnitude higher than serum concentrations. Therefore, a small amount of cellular lysis could liberate a relatively large amount of ferritin. However, nearly all serum ferritin appears to be secreted. There are probably several cell types that are sources of secreted extracellular ferritin, including iron-recycling macrophages and hepatocytes. Circulating ferritin consists almost exclusively of L-chain subunits. In contrast, intracellular ferritin contains a mixture of H and L subunits. In addition, unlike intracellular ferritin, circulating ferritin is glycosylated, thus suggesting that it passes through the endoplasmic reticulum and Golgi apparatus in a manner similar to that of other secreted proteins.
Serum ferritin levels decline with iron deficiency and rise with iron overload. In the absence of severe liver disease, infection, or chronic inflammation, serum ferritin is roughly proportional to total body iron stores. The correlation between serum ferritin levels and body iron stores is useful in the evaluation of patients with possible iron deficiency or iron overload. A low serum ferritin level (<12 µg/L) invariably represents iron deficiency, and high serum ferritin levels are found in patients with iron overload. Extremely high serum ferritin levels should be interpreted cautiously, however, because the correlation between ferritin levels and body iron stores is approximately linear only for storage reserves of iron ranging between 1 and 3 g. In addition, normal circulating ferritin values vary with sex and age. These considerations must be factored into any evaluation of iron stores based on ferritin values, particularly in children.
Various conditions modify serum ferritin levels. Inflammation increases the serum ferritin concentration severalfold. Infections, particularly chronic conditions such as tuberculosis or osteomyelitis, may also increase levels substantially, presumably because of the associated inflammatory response. Autoimmune disorders, chronic renal disease, and chronic liver disease are likewise associated with elevated levels. A number of tumors are variably associated with an increased level of circulating ferritin. For example, ferritin is an important prognostic factor in childhood neuroblastoma, in which serum ferritin levels correlate with disease severity.
The mechanisms by which inflammation and tumors increase the quantity of plasma ferritin are not fully understood. Treatment with interleukin-1β, a prime mediator of the inflammatory response, increases synthesis of ferritin in human hepatoma cells, whereas tumor necrosis factor has been shown to increase ferritin mRNA levels in murine cells in culture. These cytokines may also increase ferritin synthesis in cells secreting the protein. In addition, the inflammatory cytokine interleukin-6 induces hepcidin expression, which results in increased macrophage iron storage and probably contributes to increased serum ferritin levels. Furthermore, serum ferritin elevated out of proportion to iron stores and the transferrin saturation is a hallmark feature of ferroportin disease (see later).
Soluble transferrin receptors are also found in serum. Small vesicles containing transferrin receptors are shed from reticulocytes during their maturation to erythrocytes. In addition to these vesicle-associated receptors, the extracellular portion of the transferrin receptor lacking its transmembrane anchor can be found in the circulation. One mechanism by which soluble transferrin receptors are generated is a membrane-associated protease activity that clips the molecule between amino acids 100 and 101, at the base of its extracellular stem. This cleavage is potentiated by mutation of an O -linked glycosylation site at amino acid 104, thus suggesting that differential glycosylation may play a regulatory role. The transferrin-binding domain is intact in these soluble receptors, and they are likely to be complexed with transferrin in serum.
Because maturing red cells shed their transferrin receptors, the amount of soluble transferrin receptor in plasma reasonably reflects the degree of erythropoiesis. Measurement of plasma levels of soluble transferrin receptor provides a means of estimating erythropoietic activity that is simpler than the relatively cumbersome PIT determination. Soluble transferrin receptor (sTfR) is present in substantially lower amounts in patients with aplastic anemia relative to normal individuals. In contrast, values in patients with anemia caused by ineffective erythropoiesis are markedly increased. Patients with iron deficiency also have increased levels of circulating soluble transferrin receptors. This may result in part from the increase in cellular transferrin receptor expression produced by iron starvation and in part from the increased erythroid turnover associated with the ineffective erythropoiesis of iron deficiency. sTfR and serum ferritin values can be considered together as the ratio of sTfR to the log of ferritin (sTfR-F index). Values greater than 1.5 suggest iron deficiency alone or in combination with an inflammatory condition; values less than 1.5 are characteristic of the anemia of chronic inflammation. The sTfR-F index also appears to be sensitive enough to detect iron deficiency before iron-restricted erythropoiesis is clinically apparent. sTfR and the sTfR-F index are both decreased in patients with iron overload.
Extremes of Iron Balance
Iron disorders are invariably abnormalities in iron balance or distribution, or both. Iron deficiency is primarily due to acquired causes. In contrast, primary iron overload usually results from genetic abnormalities that perturb the regulation of intestinal iron absorption. Disorders affecting other steps in transport may be manifested as inappropriate iron accumulation in some tissues and iron deficiency in other tissues.
Iron Deficiency
Epidemiology of Iron Deficiency
Iron deficiency is the most frequent and widespread nutritional deficiency in the world because it is common in developing and developed countries alike. In fact, iron deficiency is the only micronutrient deficiency that is prevalent in virtually all developed countries. To this end, one of the U.S. national health objectives for 2010 was to reduce iron deficiency in vulnerable populations such as toddlers and women of childbearing age by 3% to 4%.
Increasing rates of breast-feeding and the availability of iron-fortified formula (see later), in conjunction with initiatives such as the U.S. Special Supplemental Food Program for Women, Infants and Children and the American Academy of Pediatrics’ promotion of formula in place of cow’s milk, have greatly reduced the prevalence of iron deficiency anemia in infants in developed countries. Nonetheless, iron deficiency, both with and without anemia, remains relatively common. According to the Fourth National Health and Nutrition Examination Survey (NHANES IV), iron deficiency without anemia exists in 7% of toddlers aged 1 to 2 years, 9% of adolescent girls, and 16% of women of childbearing age. Adolescents participating in strenuous training are another pediatric subpopulation at risk for iron deficiency. For example, young military recruits and elite adolescent athletes have an increased risk of nonanemic iron deficiency.
Socioeconomic factors are associated with iron deficiency anemia in children. For example, infants and children of low-income and minority backgrounds have higher documented rates of iron deficiency anemia. Although food insecurity is known to be associated with iron deficiency anemia, other factors such as bottle-feeding patterns may also play a role in the prevalence of iron deficiency. The iron status of young children correlates closely with the iron status of their mothers, thus indicating that a constellation of factors in the child’s environment influence iron intake and therefore iron status.
Phases of Development of Iron Deficiency
Because most of the body’s iron is directed toward synthesis of hemoglobin, erythrocyte production is among the first casualties of iron deficiency to become clinically apparent in usual laboratory evaluations. However, it actually represents a late stage of iron depletion. Iron deficiency progresses through three discernible phases:
1. Prelatent iron deficiency occurs when tissue stores are depleted, without a change in hematocrit or serum iron levels. This stage of iron deficiency may be detected by low serum ferritin measurements.
2. Latent iron deficiency occurs when reticuloendothelial macrophage iron stores are depleted. The serum iron level drops and TIBC increases without a change in hematocrit. This stage may be detected by a routine check of fasting, early-morning transferrin saturation. Erythropoiesis begins to be limited by a lack of available iron, and sTfR levels increase. The reticulocyte hemoglobin content (CHr) decreases because newly produced erythrocytes are iron deficient. The bulk of the erythrocyte population appears normal. For this reason, sole reliance on indicators derived from the entire erythrocyte population frequently fails to detect this stage of iron deficiency.
3. Frank iron deficiency anemia is associated with erythrocyte microcytosis and hypochromia. It is detected when iron deficiency has persisted long enough that a large proportion of the circulating erythrocytes were produced after iron became limiting.
Etiology of Iron Deficiency
The development of iron deficiency is a result of the interaction between iron intake, physiologic iron requirements, and the potential for blood loss.
Inadequate Iron Intake
Dietary Sources.
Heme, derived from animal tissues, is the most readily absorbed form of iron. Uptake occurs independently of gastric pH and, like the uptake of nonheme iron, is increased in patients with high marrow erythroid activity. Much of the world’s population eats little or no meat; for these peoples, nutrition is derived from cultivated grasses such as rice, plants that are relatively poor sources of iron. Eating such a diet is one of the factors that make iron deficiency anemia the most common nutritional anemia worldwide.
In developing countries, nutritional iron deficiency is often compounded by chronic blood loss from parasitic infections and malaria (see later). In industrialized countries, however, iron deficiency is usually due to insufficient dietary intake to meet physiologic needs. Because of their rapid growth and increased need for iron, infants, toddlers, female adolescents, and pregnant women are especially vulnerable.
Consumption of cow’s milk may contribute to iron deficiency through several mechanisms. Cow’s milk and human milk both have low iron content, but the bioavailability of iron in human milk is greater. Therefore, the transition from human milk to cow’s milk may place toddlers at risk for iron deficiency. Indeed, the prevalence of iron deficiency increases with the duration of bottle-feeding of cow’s milk. Cow’s milk also compounds iron deficiency by replacing iron-rich foods in the diet. In addition, components of cow’s milk, such as calcium and caseinophosphopeptide, can directly interfere with iron absorption. Furthermore, whole cow’s milk contains proteins that may irritate the lining of the gastrointestinal tract in infants, and even low-grade but chronic hemorrhage may produce significant iron deficiency. The neonatal growth spurt requires a tremendous quantity of iron, thus compounding the disadvantages of cow’s milk. For these reasons, current recommendations exclude cow’s milk from the infant’s diet in the first year of life, limit subsequent consumption of cow’s milk to 24 ounces per day, suggest that nonbreastfed infants should receive iron-fortified formulas, and advise an additional source of iron for infants who are breastfed after 4 to 6 months of age, when they have depleted the excess iron that was present in newborn stores and erythrocytes.
Poor Bioavailability.
Although iron is abundant in the environment, it is nearly insoluble in aqueous solution, which makes acquisition of the common element a major challenge. Most environmental iron exists as insoluble salts. Gastric acidity assists in conversion to absorbable forms, but the efficiency of this process is limited. Many plant products contain iron, but absorption frequently is limited both by low solubility and by powerful natural chelators that bind ambient iron. Phytates (organic polyphosphates) found in wheat products, for example, bind iron with tremendous avidity. The challenge of acquiring sufficient iron from the environment may have been an important factor in the spread of genes for hemochromatosis disorders.
High gastric pH reduces the solubility of inorganic iron and thereby impedes absorption. Surgical interventions, such as vagotomy or hemigastrectomy for peptic ulcer disease, formerly were the major causes of impaired gastric acidification with secondary iron deficiency, particularly in adults. The histamine H 2 blockers and the more recently introduced acid pump blockers, used to treat peptic ulcer disease and acid reflux, may cause defective iron absorption. There is, however, little evidence that they substantially alter iron status in those without preexisting iron deficiency.
The impaired function of gastric parietal cells associated with pernicious anemia not only reduces the production of intrinsic factor but also lessens gastric acidity and hence compromises absorption of iron. In addition, megaloblastic enterocytes absorb iron poorly. Frank iron deficiency can accompany the anemia produced by cobalamin deficiency. However, the rarity of pernicious anemia in children makes this complication uncommon in pediatric populations.
A number of environmental factors can produce dietary iron deficiency by interfering with iron absorption, including metals such as cobalt, which share the iron absorption machinery. Lead may fall into this category, although it remains to be definitively established whether DMT1 is the primary intestinal lead transporter in vivo. Regardless of the mechanism, it is clear that iron deficiency increases the rate of uptake of both iron and lead from the gastrointestinal tract. Iron deficiency and lead intoxication frequently coexist.
Malabsorption.
Some disorders disrupt the integrity or surface area of the enteric mucosa and thereby hamper iron absorption ( Box 11-2 ). Consequently, in the absence of blood loss (see below), iron deficiency unresponsive to oral iron therapy should prompt an investigation for intestinal diseases that affect the duodenum. Inflammatory bowel disease, particularly Crohn disease, may injure extensive segments of the small intestine, including the jejunum and duodenum. Invasion of the submucosa by inflammatory cells and disruption of the tissue architecture impair iron absorption and uptake of dietary nutrients. Occult gastrointestinal bleeding exacerbates the problem. The result is iron deficiency anemia complicated by the anemia of chronic inflammation. Furthermore, Crohn disease frequently involves the terminal ileum, with the concurrent development of cobalamin deficiency. These disorders are not diagnostic enigmas. Patients with extensive bowel involvement are very ill. Diminishing the underlying inflammatory process best treats the iron deficiency.
INADEQUATE ABSORPTION
Poor bioavailability (absorption of heme Fe > Fe 2+ > Fe 3+ )
Antacid therapy/high gastric pH
Bran, tannins, phytates, starch
Other metals (e.g., cobalt, lead)
Loss or dysfunction of absorptive enterocytes (e.g., gluten-sensitive enteropathy, gastric bypass, Crohn disease)
INSUFFICIENT OR INACCESSIBLE IRON STORES
Physiologically Increased Requirements
Pregnancy, particularly repeated
Rapid growth (e.g., infancy)
Gastrointestinal Blood Loss
Epistaxis
Gastritis
Ulcer
Meckel diverticulum
Milk-induced enteropathy
Parasitosis
Varices
Tumor or polyps
Inflammatory bowel disease
Arteriovenous malformation
Colonic diverticula
Hemorrhoids
Vaginal Blood Loss
Increased menstrual blood loss
Tumor
Urinary Blood Loss
Chronic intravascular hemolysis (e.g., paroxysmal nocturnal hemoglobinuria)
Chronic glomerulonephritis
Chronic infection
Tumor
Pulmonary Blood Loss
Pulmonary hemosiderosis
Tuberculosis
Bronchiectasis
Inflammation or Infection
Defects in intestinal iron uptake (e.g., TMPRSS6 mutations)
INADEQUATE PRESENTATION TO ERYTHROID PRECURSORS
Atransferrinemia
Anti–transferrin receptor antibodies
ABNORMAL INTRACELLULAR TRANSPORT OR UTILIZATION
Erythroid iron-trafficking defects (e.g., DMT1 mutations)
Defects in heme biosynthesis
Tropical sprue and celiac disease (gluten-sensitive enteropathy) can also interfere with iron absorption. Loss of the villous absorptive surface along with chronic inflammation causes profound malabsorption, although some patients with deranged iron absorption lack gross or even histologic changes in the structure of the bowel mucosa. The anemia in these patients is often complicated by a superimposed, generalized nutritional deficiency. Celiac disease frequently improves with ingestion of a gluten-free diet, with secondary correction of the anemia.
Iron absorption may be disrupted when substantial segments of bowel, particularly the proximal duodenum, are removed surgically. Intractable inflammatory bowel disease, traumatic abdominal injury, and structural complications such as intestinal volvulus or intussusception all may necessitate intestinal resection. Likewise, obesity surgeries that bypass the proximal duodenum may limit iron absorption. Iron deficiency develops slowly afterward and may not become evident for several years after the surgical procedure.
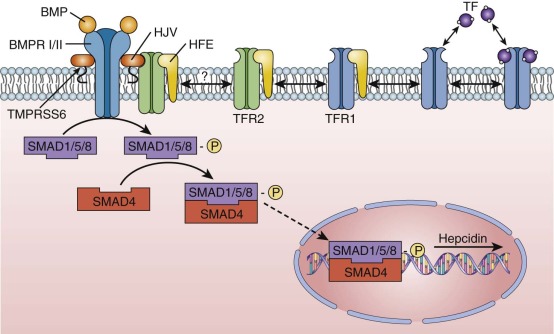
Malabsorption of iron is rare in the absence of gross or microscopic structural defects of the intestine or the anemia of chronic inflammation (see later). However, multiple families with an autosomal recessively inherited predisposition to iron deficiency and systemic iron malutilization have been described. These individuals exhibit iron deficiency anemia at an early age that is characterized by a nearly complete lack of response to oral iron supplementation and a slow, incomplete response to parenteral iron therapy—so-called iron-refractory, iron deficiency anemia (IRIDA). Individuals with IRIDA have biallelic inactivating mutations in the TMPRSS6 protein, a transmembrane serine protease expressed in the liver, that leads to inappropriate hepcidin overexpression and eventuates in impaired iron absorption and iron recycling ( Fig. 11-6 ). Importantly, a common single-nucleotide polymorphism (SNP) in the protein-coding region of TMPRSS6 (a substitution of a valine for an alanine at amino acid position 736) is strongly associated with lower plasma iron and ferritin levels as well as several red blood cell parameters, and has been shown to modify the severity of disorders of iron overload. Although mutations in DMT1 might also be expected to, and in model organisms do indeed, lead to a somewhat similar congenital iron deficiency anemia phenotype as mutations in TMPRSS6; in fact, reported patients with DMT1 mutations have iron-restricted erythropoiesis in the setting of systemic iron overload.
Systemic Iron Loss
Gastrointestinal Blood Loss.
Blood loss is the leading cause of iron deficiency worldwide. The gastrointestinal tract is both the site of iron uptake and the most common site of blood loss, particularly when bleeding is not readily apparent. However, bleeding from other sites should also be considered if iron deficiency has not otherwise been explained.
Anatomic defects of the gastrointestinal tract commonly cause blood loss and consequent iron deficiency. The most frequent congenital defect is Meckel diverticulum, which results from a persistent omphalomesenteric duct. This abnormality can produce abdominal pain and, occasionally, intestinal obstruction in young children. Meckel diverticulum may equally be associated with clinically overt or occult blood loss and secondary iron deficiency. Peptic ulcer disease is extremely uncommon in children but should be considered as a cause of gastrointestinal blood loss when the clinical setting suggests it.
Other structural defects of the gastrointestinal tract may cause bleeding as well. Arteriovenous malformations involving the superficial blood vessels occur in patients with hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome). These defective vessels frequently bleed to a degree that engenders iron deficiency. Although the disorder is transmitted as an autosomal dominant trait, the characteristic vascular lesions rarely attain clinical significance before young adulthood. The condition is easily recognized because the mucosal lining of the oropharynx and nasal cavity exhibits characteristic telangiectasias.
The leading cause of gastrointestinal blood loss worldwide is parasitic infestation. Hookworm infection, caused primarily by Necator americanus or Ancylostoma duodenale , is endemic in much of the world and is often asymptomatic. Microscopic blood loss leads to significant iron deficiency, particularly in children. More than 1 billion people, most in tropical or subtropical areas, are infested with parasites. Combined, their total daily blood loss exceeds 11 million liters. The larvae spawn in moist soil and penetrate the skin of unprotected feet. Hookworm infection, once prevalent in the southeastern United States, declined precipitously with better sanitation and routine use of footwear when outside.
Trichuris trichiura , the culprit in trichuriasis or whipworm infection, is believed to infest the colon of 600 to 700 million people. Only 10% to 15% of these individuals have worm burdens sufficiently great to produce clinical disease. However, most are children between the ages of 2 and 10 years. Growth retardation, in addition to iron deficiency, occurs with heavy infestation. Trichuriasis is the most common helminthic infection encountered in Americans returning from visits to tropical or subtropical regions of the world.
Urinary Blood Loss.
Occasionally, blood loss into the urinary tract outstrips iron absorption. However, urinary blood loss is usually apparent and sufficiently alarming that patients seek medical attention before substantial iron deficiency develops. Iron deficiency resulting from hematuria secondary to renal disease is relatively uncommon.
The best characterized cause of significant renal blood loss is Berger disease, which produces relapsing episodes of gross or microscopic hematuria. The disorder occurs most commonly in older children and young adults. It is characterized by diffuse mesangial proliferation or focal and segmental glomerulonephritis. Diffuse mesangial deposits of IgA are the hallmark of the disorder. Although the disease spontaneously remits in some children, progression to end-stage renal disease may occur. Occasionally, Goodpasture syndrome produces substantial urinary blood loss. Immunofluorescent staining of biopsy specimens reveals the characteristic antibodies to basement membrane lining the glomeruli. Blood loss into the urinary bladder can occur in association with infectious cystitis. Hematuria to the point of iron deficiency is extremely uncommon, however.
Patients with chronic intravascular hemolysis, such as those with paroxysmal nocturnal hemoglobinuria or arterial jet lesions from congenital or acquired heart disease with or without endovascular appliances, may also develop significant renal blood loss.
Pulmonary Blood Loss.
Although pulmonary blood loss may be sufficiently severe to produce iron deficiency, it is distinctly rare. Idiopathic pulmonary hemosiderosis primarily affects children and young adolescents (reviewed by Specks ). Any child with iron deficiency and unexplained pulmonary disease should be evaluated for this potentially deadly disorder, which is characterized by slow but intractable hemorrhage into the bronchioles and alveoli. Iron deficiency anemia is a major part of the clinical picture of pulmonary hemosiderosis inasmuch as pulmonary macrophages are distinct from the normal iron-recycling circuit and iron trapped within these cells is effectively lost from the functional systemic pool. Consequently, a paradoxic situation evolves in which iron deficiency anemia develops in the presence of a surfeit of total body iron. Bronchoalveolar lavage reveals hemosiderin-laden macrophages that are characteristic of the disorder. Gastric aspiration, which is a less invasive procedure, may also reveal these cells due to swallowing of sputum. Chronic pulmonary infection with bronchiectasis, once considered to be a frequent cause of pulmonary hemorrhage, is now rare. Iron that accumulates in these cells induces the formation of toxic products, which leads to the activation of macrophages. These cells begin to damage the delicate lining of the bronchoalveolar tree and produce severe fibrosis. Oxygen exchanges poorly across the damaged alveolar surfaces, thereby lowering the efficiency of oxygen/carbon dioxide exchange. Late in the course of pulmonary hemosiderosis, the radiographic picture of the lungs is striking and frequently shows marked retraction and scarring. The restrictive lung disease and substantial pulmonary arterial shunting across the lung may eventually be fatal.
Most often the cause of pulmonary hemosiderosis evades elucidation. In a number of patients, it occurs in conjunction with disorders of immune dysfunction, including celiac disease and Goodpasture syndrome. Treatment of the associated disorder can lead to remission of the pulmonary process, consistent with an immunologic mechanism of the lung injury. Chloroquine treatment has been used with modest success in some patients. Cyclophosphamide has produced responses in a number of patients. A combination of prednisone and azathioprine has also been used. Other immunosuppressive agents may prove to be even more effective as immunologic suppressers in the treatment of this condition, but the infrequency of the disorder means that a large, multicenter trial is needed to collect definitive treatment information. Although the prognosis was once believed to be very poor for all patients with idiopathic pulmonary hemosiderosis, more recent data suggest that such may not be the case.
Menstrual Blood Loss.
Blood loss associated with menstruation predisposes women to iron deficiency. In a sample of healthy, menstruating, nonpregnant Danish women 18 to 30 years of age not receiving iron supplementation, serum ferritin levels correlated inversely with the duration and intensity of menstrual bleeding. Menorrhagia or dysfunctional uterine bleeding may lead to frank iron deficiency anemia. In many cases, dysfunctional uterine bleeding is due to anovulation, a common phenomenon in pubertal females. Progestational agents or oral contraceptives may ameliorate the dysfunctional uterine bleeding or menorrhagia. If this approach does not resolve the abnormal bleeding, other causes such as pelvic abnormalities or disorders of hemostasis, particularly von Willebrand disease, should be considered. Women with menorrhagia, especially menorrhagia present since menarche, have a high incidence of von Willebrand disease (13%), which in the general population is 1% to 2%.
Consequences of Iron Deficiency
Although anemia is the most prominent manifestation of iron deficiency, there may be other clinically significant sequelae of iron deficiency, even in the absence of anemia. Organ and tissue dysfunction, including impaired immunity, decreased muscle performance and neurocognitive function, and poor weight gain, may occur with iron deficiency. Significant inroads in understanding some of these effects, particularly cognitive dysfunction in young children, have been made in the past few years.
Neurocognitive Effects.
The association between iron deficiency anemia and impaired neurocognitive function is well established, and this association holds even when potential confounders such as psychosocial and environmental factors are taken into account. Infants and toddlers, who are undergoing critical neurocognitive development, may be at particular risk for such effects.
Iron deficiency that has not yet progressed to anemia is also associated with impaired mental and motor functioning. For example, when tested with the Bayley Scales of Infant Development, nonanemic iron-deficient infants had lower developmental scores than iron-sufficient infants did, and these abnormalities were detected in children as young as 9 to 12 months. In addition, the degree of developmental deficits in infants may correlate with the level of iron deficiency. Older populations of children, such as nonanemic iron-deficient preschoolers, have lower mental development scores, and school-age children and adolescents have lower math and verbal scores than their iron-sufficient peers do.
In some investigations, dietary iron supplementation has reversed the cognitive dysfunction. For example, in the study of 9- to 12-month-old infants cited earlier, iron replacement increased Mental Development Index scores substantially in only 7 days. Numerous other studies, however, failed to show improvement in depressed performance. Lozoff and colleagues have also demonstrated that these effects remain evident as long as 19 years later and that children of low socioeconomic status may be disproportionately affected. Older patients with iron deficiency may express a subjective state of cognitive impairment, likely reflecting central nervous system iron deficiency, that is rapidly reversed by treatment.
Iron deficiency may increase the risk for lead exposure through either pica or increased absorption. Concomitant lead toxicity can further hamper the psychological development of these children. Information on the long-term effects of iron deficiency during infancy highlights the importance of early detection and intervention. Health care providers must therefore work actively to prevent and detect iron deficiency during infancy.
The mechanism by which iron deficiency impairs neurologic function is unknown. Iron deficiency could impair neurotransmitter mechanisms. It has been shown to decrease expression of dopamine receptors in the rat brain. Iron deficiency may also interfere with myelination and alters myelin proteins and lipids in oligodendrocytes. In addition, several enzymes in neural tissue require iron for normal function. The cytochromes involved in energy production, for example, are predominantly heme proteins.
Pica.
Pica, the compulsive consumption of nonnutritive substances, is a recurrent symptom in patients with iron deficiency. The precise pathophysiology of pica is unknown, but it is probably attributable to central nervous system iron deficiency. Patients often consume starch, ice, soil, foam, or clay. Both clay and starch can bind iron in the gastrointestinal tract and exacerbate the deficiency. A dramatic example of the problems produced with clay consumption occurred in the 1950s in iron-deficient children along the border between Iran and Turkey. These youngsters had other peculiar abnormalities, including massive hepatosplenomegaly, poor wound healing, and a bleeding diathesis. Presumably, the children initially had simple iron deficiency associated with pica, including geophagia. The soil contained compounds that bound both iron and zinc. Secondary zinc deficiency was thought to cause the hepatomegaly and other unusual features.
Epithelial Changes.
Iron deficiency produces significant gastrointestinal tract abnormalities, reflecting the enormous proliferative capacity of this tissue. Angular stomatitis and glossitis with painful swelling of the tongue may develop. The flattened, atrophic, lingual papillae make the tongue smooth and shiny. A rare complication of iron deficiency is Plummer-Vinson syndrome, with the formation of a postcricoid esophageal web. Long-standing, severe iron deficiency affects the cells that generate the fingernails and produces koilonychia, or spooning. The nail substance is soft, so ordinary pressure on the fingertips, as occurs with writing, for instance, produces a concave deformity. Most of these abnormalities are now uncommon in industrialized nations and are particularly rare in children even given the relative frequency of iron deficiency.
Iron Deficiency Anemia.
On physical examination, pallor, tachycardia, splenomegaly and a systolic murmur may be appreciated. As is true of all anemias, the anemia of iron deficiency impairs tissue oxygenation and produces the symptoms of pallor, weakness, fatigue, and light-headedness. The morphologic features of the peripheral blood smear in patients with iron deficiency anemia vary greatly according to the severity and duration of the condition ( Fig. 11-7 ). The earliest morphologic features of iron-deficient erythropoiesis in the peripheral blood are subtle variations in red blood cell size and the degree of hemoglobinization, with occasional smaller, less well hemoglobinized cells recognizable. These parameters are reflected by changes in the red cell distribution width (RDW) and the hemoglobin distribution width (HDW) and are usually present before overt hypochromia and microcytosis are seen. Indeed, at this point, the hemoglobin and the mean cell volume (MCV) may be in the normal range. As the severity and duration of iron-deficient erythropoiesis increase, so do the extent of morphologic changes, with increasing variation in cell size (anisocytosis) and hypochromia and distinctive misshapen hypochromic microcytes with irregular, often angulated borders present (poikilocytes). The plasma membranes of iron-deficient red cells are abnormally stiff, and such cells are more prone to hemolysis. This rigidity may contribute to poikilocytic changes, seen particularly with severe iron deficiency. Small, stiff, misshapen cells are cleared by the reticuloendothelial system, thereby further contributing to low-grade hemolysis. The cause of this alteration in membrane fluidity is unknown. Very thin, elongated hypochromic cells, so-called pencil cells, are said to be characteristic of iron deficiency. Occasional target cells, in which hemoglobin pools in the center of the erythrocyte, may be seen, but these cells, as well as polychromasia, indicative of a reticulocytosis, are not as prevalent as in β-thalassemia. Basophilic stippling, nucleated red blood cells, or nuclear remnants (i.e., Howell-Jolly bodies) are generally not present, distinguishing severe iron deficiency from thalassemias or hemoglobinopathies. β-thalassemia trait (see Chapter 21 ) also produces microcytic cells and borderline anemia and is sometimes confused with early iron deficiency. Iron deficiency, however, alters red cell size unevenly, resulting in a high RDW, which is typically normal in β-thalassemia trait. Other common features of β-thalassemia trait are rare cells with basophilic stippling and target cells, which are generally not present in the earliest phases of iron deficiency. These characteristics are not sufficiently distinctive to be diagnostically definitive, however, and any suspicion should be confirmed with more objective tests such as a hemoglobin electrophoresis. Importantly, iron deficiency may lead to an elevated hemoglobin A2, confounding the interpretation of hemoglobin electrophoresis results. Given the not uncommon coexistence of iron deficiency and β-thalassemia trait, any abnormal hemoglobin electrophoresis should be interpreted in the context of iron studies (see below) and repeated after iron supplementation in the iron-deficient patient.
Thrombocytosis with platelet counts in the range of 500,000 to 700,000 cells/fL occurs frequently in iron-deficient patients. Megakaryocytes and normoblasts are derived from a common committed progenitor cell, the colony-forming unit–granulocyte, erythrocyte, macrophage, megakaryocyte (CFU-GEMM). Thrombopoietin, the molecule that stimulates the growth of megakaryocytes and the production of platelets, is homologous to erythropoietin (see Chapter 6 ). The high levels of erythropoietin produced by iron deficiency anemia conceivably could cross-react with megakaryocyte thrombopoietin receptors and modestly raise the platelet count. However, this hypothesis has not been proved experimentally.
Bone marrow examinations are no longer routinely used to assess iron stores. However, when they are performed, in severe cases, late erythroblasts often have ruffled or frayed edges, not unlike other hypochromic anemias. Iron staining is best performed on aspirate smears rather than biopsies, as decalcification can leach iron out of the sample, resulting in false-negative results, and the cytologic preservation is typically inadequate to resolve minute, intraerythroid iron granules (sideroblasts, see below) indicative of erythroid iron sufficiency. Iron-stained marrow particles should have readily identifiable cells with coarse iron positivity, representing macrophages with abundant ferritin and hemosiderin storage iron. If particles are absent, careful microscopic examination of normal, iron-replete bone marrows under oil immersion should demonstrate up to 30% to 40% of the erythroid precursors with several fine, peripherally placed iron deposits that represent iron in cytosolic ferritin. Such cells are termed sideroblasts , are entirely normal, and are absent or decreased in iron deficiency. In the anemia of inflammation, where there is a defect in iron recycling, macrophage iron stores may be present but intraerythroblast iron will be decreased or absent (see below).
Diagnosis of Iron Deficiency
Because they are inexpensive and widely available, hematologic markers are frequently used to assess iron status. A hemoglobin content less than the fifth percentile in a reference population is often used to define anemia. Changes in erythrocyte parameters accompanying iron deficiency anemia include a decrease in MCV, reflecting microcytosis, and an increase in RDW, reflecting anisocytosis, as described earlier. Because these parameters represent averages of the entire red blood cell population, iron deficiency must be present for some time before it deviates from these measures.
The American Academy of Pediatrics and the U.S. Centers for Disease Control and Prevention recommend the use of hemoglobin to screen children at risk for iron deficiency. Because only the later stages of iron deficiency result in anemia, relying on hemoglobin (or hematocrit) to screen for iron deficiency misses many children who are, in fact, iron deficient and in whom adverse consequences, such as potentially irreversible neurocognitive impairment, may have already begun to occur. For example, based on NHANES data, a hemoglobin concentration of less than 11 g/dL has a sensitivity of 30% for detecting iron deficiency, as defined by ferritin, erythrocyte protoporphyrin, and transferrin saturation (see below). That adverse consequences of iron deficiency may occur in the absence of anemia highlights the importance of diagnostic approaches that detect iron deficiency early, without relying on hematologic indicators that are altered only after iron deficiency has progressed.
Serum or plasma biochemical parameters based on iron metabolism are often used to diagnose iron deficiency. The tests that are commonly employed, as well as their physiologic relevance and confounding variables, are listed in Table 11-1 . Plasma iron concentrations, largely reflecting the iron recycled from effete red blood cells, fall as iron is depleted, but they are also affected by diurnal variation and dietary intake. The availability of plasma iron-binding sites, or TIBC, increases as iron stores fall. Serum iron concentration divided by TIBC yields the transferrin saturation, which is expressed as a percentage of transferrin iron-binding sites occupied by iron. A transferrin saturation of less than 10% is a stringent definition of iron deficiency that is often employed as the “gold standard” against which other tests are judged. However, erythropoiesis is to some extent iron restricted at a transferrin saturation of 15% or less. Serum ferritin is a glycosylated form of the intracellular iron storage protein L-ferritin that is incapable of binding iron. The precise ontogeny and cells of origin of this circulating form of the protein are uncertain. Nevertheless, the serum ferritin is a good biomarker of total body iron stores and is roughly linearly related to this parameter over a wide range—from iron deficiency to moderate iron overload. A low serum ferritin level is a very specific indicator of iron deficiency, but ferritin is an acute-phase reactant and is elevated in chronic inflammatory states, independent of body iron status, thus reducing the sensitivity of the test. Ferritin has in fact been shown to be a poor indicator of iron deficiency in the pediatric population. Whereas ferritin is a “positive” acute-phase reactant and may lead to an overestimate of iron stores in the setting of inflammation, serum iron and TIBC are “negative” acute-phase reactants and are reduced in this context.
| Test | What It Indicates | Result in Iron Deficiency | Result in Iron Overload | Confounding Conditions |
|---|---|---|---|---|
| Iron | Iron recycling/usage, stores (indirect) | ↓ | ↑-↑↑ | ↓ in inflammation |
| Total iron-binding concentration (TIBC) | Serum/plasma transferrin | ↑ | Normal | ↓ in inflammation |
| Transferrin saturation | % plasma iron-binding sites on transferrin that are occupied | ↓-↓↓ | ↑-↑↑ | ↓ in inflammation |
| Free or zinc protoporphyrin | Functional mitochondrial iron status | ↑ | Normal | ↑ in reticulocytosis |
| Soluble transferrin receptor (sTfR) | Erythropoietic mass | ↑ | Normal | Ineffective erythropoiesis |
| Ferritin | Iron stores (indirect) | ↓-↓↓ | ↑-↑↑ | ↑ in inflammation |
| Bone marrow iron | Macrophage iron stores/retention, red blood cell iron uptake | ↓ | Normal to ↑ | ↑ in inflammation |
| Liver iron | Iron stores | ↓-↓↓ | ↑-↑↑ | Secondarily increased in cirrhosis |
| Hepcidin † | Hepatocyte iron | ↓-↓↓ | ↑-↑↑ | ↑ in inflammation |
* All measures are in serum or plasma unless otherwise indicated.
† Hepcidin is not yet routinely available as a clinical test.
Because of the limitations of both hematologic and biochemical tests, novel methods of diagnosing iron deficiency have been sought. Measures of red blood cell protoporphyrin IX, particularly zinc protoporphyrin (ZPP), assess the functional availability of iron to the erythrocyte mitochondrion, where iron is incorporated into protoporphyrin IX by ferrochelatase to form heme. In the context of mitochondrial iron insufficiency, zinc, an alternative ferrochelatase substrate, is inserted into the protoporphyrin ring. Measurement of the absolute level of red blood cell ZPP or, more specifically, the ratio of ZPP to iron protoporphyrin IX (i.e., heme), which is increased in iron deficiency, therefore provides an assessment of delivery of iron to erythroid mitochondria for heme synthesis.
sTfR is a truncated form of the transferrin receptor that is cleaved from reticulocytes and erythroid cells and circulates in plasma largely bound to transferrin. sTfR is increased in iron deficiency but is also increased in other anemias characterized by ineffective erythropoiesis (e.g., thalassemia). Though not uniformly available in all clinical laboratories, it may be a particularly important adjunct in distinguishing iron deficiency anemia from the anemia of inflammation, in which it is not increased.
The cellular hemoglobin of the reticulocyte (CHr) is another test available only in selected laboratories because it is unique to instruments manufactured by Bayer (Tarrytown, NY). CHr has been evaluated prospectively as a means of routinely screening infants and toddlers. In this setting, a CHr cutoff of 27.5 pg had 83% sensitivity and 72% specificity, whereas a hemoglobin concentration of less than 11 g/dL was a poor predictor of iron deficiency, with only 26% sensitivity. CHr has also been used to accurately assess the iron status of potential adult blood donors. A summary of the relative sensitivity and specificity and approximate cost of each of the tests commonly employed to screen for iron deficiency is shown in Table 11-2 .
| Method | Age | Cost | Sensitivity | Specificity | Reference |
|---|---|---|---|---|---|
| Transferrin saturation (<10%) | any | ≈$85 | 100% | 100% | “Gold standard” defined as 100% sensitive and specific |
| Hb (<11 g/dL) | 9-12 months | ≈$30 | 26% | 95% | |
| Hb (<10.7 g/dL) | 9-12 months | ≈$30 | 67% | 87% | |
| MCV (<71 fL) | 9-12 months | ≈$30 | 86% | 83% | |
| CHr (<27.5 pg) | 9-12 months | ≈$50 | 83% | 72% | |
| Spun Hct (<33%) | 9-36 months | ≈$15 | 16% | 92% | |
| Ferritin (<10 µg/L) | 4-15 months | ≈$65 | 71% | 57% | |
| Ferritin (<10.9 µg/L) | 9-12 months | ≈$65 | 83% | 80% | |
| ZPP (>50 nmol/mol heme) | 9-36 months | ≈$150 | 81% | 88% | |
| FEP (>2.12 µmol/L RBC) | 4-15 months | ≈$150 | 82% | 57% | |
| sTfR (>13.5 mg/L) | 4-15 months | ≈$90 | 31% | 95% |
Although commercial tests for urinary or plasma hepcidin are not yet currently in clinical use, measurement of this analyte may well have a role in assessing iron status, whether it be deficiency or overload, in the future. This will certainly be the case if algorithms for determining if the hepcidin is appropriate, or inappropriately high or low, for the clinical context, permitting some insight into the pathophysiology of the disorder under investigation. However it is ultimately implemented, analysis of hepcidin levels will, similar to the serum ferritin, be confounded by concomitant inflammation, or, like sTfR, ineffective hematopoiesis.
Treatment of Iron Deficiency
The most important steps in the evaluation and treatment of iron deficiency are determining the cause and correcting the abnormality. Malignant disease of the gastrointestinal tract, the most worrisome cause in adults with iron deficiency, is vanishingly rare in children. Whereas in children, growth spurts, poor dietary patterns, menstrual losses, and non-malignant causes of gastrointestinal bleeding are more often the cause of iron deficiency. After initial diagnostic investigations, oral iron supplementation usually replaces stores most efficiently.
Oral Supplementation.
Iron salts offer inexpensive, effective therapy for iron deficiency. Although ferrous sulfate is most often recommended, patients frequently complain of gastrointestinal discomfort, constipation, and bloating, as well as stool discoloration, thus making its use unacceptable to many. Interestingly, in a randomized, controlled trial comparing 3 mg/kg/day of ferrous sulfate drops with placebo in 278, 12-month-old infants, there was no difference in the frequency of vomiting, diarrhea, or fussiness, and infants in the placebo group had a higher incidence of constipation (95% vs. 1%). Iron is frequently provided to children at a dose of 3 mg/kg/day divided into three-times-a-day dosing. Single-dose daily regimens, however, are tolerated as well as three-times-a-day regimens. Administration of iron on an empty stomach at night will lessen the gastrointestinal difficulties. The decreased gastrointestinal motility of sleep will also enhance absorption.
The absorptive capacity of the normal duodenum for iron is essentially saturated with about 25 mg of elemental iron in ionic form. Ferrous gluconate, which is roughly equivalent in cost, contains about 50 mg of elemental iron per tablet. This form of replacement may produce fewer problems than ferrous sulfate and is excellent as the initial treatment of iron deficiency. Ascorbic acid supplementation enhances iron absorption, although it has a relatively minor effect in individuals ingesting normal, balanced diets. Combination tablets containing iron salts and ascorbic acid are significantly more expensive than separate tablets for each. Even with faithful use of oral iron, adequate replacement of body stores in patients with moderate iron deficiency anemia requires several months. With ongoing blood loss, replacement of stores with oral iron becomes very difficult.
Polysaccharide/iron complexes differ from iron salts. Polar oxygen groups in the sugars form coordination complexes with iron atoms. The well-hydrated microspheres of polysaccharide iron remain in solution over a wide range of pH values. Most patients tolerate this form of iron better than the iron salts, even though the 150 mg of elemental iron per tablet is substantially more than that provided by iron salts.
Iron supplements must be stored in a childproof manner. Unintentional iron poisoning from iron supplements is a leading cause of injury related to poisoning in young children and frequently occurs when children obtain improperly stored iron.
Box 11-3 lists potential causes of a poor response to oral iron supplementation. A variety of barriers, including treatment-associated factors and system-wide barriers, contribute to the shortcomings in iron deficiency screening and correction that have been documented.
Noncompliance
Ongoing blood loss
Insufficient duration of therapy
High gastric pH
Antacids
Histamine 2 blockers
Gastric proton pump inhibitors
Inhibitors of iron absorption or utilization
Lead
Aluminum intoxication (hemodialysis patients)
Chronic inflammation
Neoplasia
Incorrect diagnosis
Thalassemia disorder
Sideroblastic anemia
Anemia of chronic inflammation
Genetic disorder of iron absorption or utilization (e.g., iron-refractory, iron deficiency anemia [IRIDA])
Parenteral Iron Replacement.
Parenteral iron is now available in the United States in four intravenous forms: iron dextran, iron gluconate, iron sucrose, and iron oxide coated with polyglucose sorbitol carboxymethylether. Only some of these products are approved specifically for use in the pediatric population. Intravenous iron therapy is indicated when (1) oral iron is poorly tolerated; (2) rapid replacement of iron stores is needed; (3) gastrointestinal iron absorption is compromised; or (4) erythropoietin therapy is necessary, particularly in renal dialysis patients. Iron dextran can also be administered by intramuscular injection, but this route is discouraged because it can be painful and leakage into subcutaneous tissue produces long-standing skin discoloration. A “Z-track” injection into the muscle minimizes the chance of subcutaneous leakage. The suboptimal muscle mass frequently associated with nutritional deficiency further complicates this mode of replacement. Intravenous infusion circumvents these problems altogether. With either route of administration, a small test dose should be given and the patient observed by a physician for 30 minutes to rule out an anaphylactic reaction to the medication. Such reactions do occur but are infrequent. The newer iron sucrose and iron gluconate preparations are believed to be less likely to cause complications, but there is less accumulated experience with these preparations, and many practitioners still use iron dextran routinely.
Ten percent to 15% of patients experience transient mild to moderate arthralgias the day after intramuscular or intravenous administration of iron dextran. Acetaminophen usually relieves the discomfort effectively. Iron dextran is generally avoided in patients with rheumatoid arthritis because it may provoke painful flares of their disease. The symptoms may result from release of inflammatory cytokines such as interleukin-1 and tumor necrosis factor. Iron dextran is cleared from the circulation by macrophages, which are probably activated to release proinflammatory peptides.
In uncomplicated cases of iron deficiency, intravenous replacement may produce subjective improvement in symptoms within a few days. Peak reticulocytosis occurs after about 10 days, and complete correction of the anemia takes 4 to 6 weeks, although the hematocrit rises sufficiently in 1 or 2 weeks to provide symptomatic relief for most patients. CHr is a very sensitive measure of response. Brugnara and colleagues have demonstrated that a rise in CHr is appreciated within 2 to 4 days of the administration of intravenous iron.
Refractory bleeding that cannot be prevented, as occurs, for example, with hereditary hemorrhagic telangiectasia, presents a management problem. Oral iron supplementation often fails to keep pace with losses. Blood transfusions replace red cells in the short term and iron in the long term, but infection and alloimmunization make such therapy unacceptable for many patients. Intermittent iron replacement with infusions of intravenous iron is the most reasonable alternative. Replacements should occur over short intervals two or three times a year, with the aim of repleting body stores. A simple formula can be used to determine the replacement dose of iron dextran:
Dose ( mL ) = 0.0442 × ( Desired Hb − Observed Hb ) × Lean body weight + ( 0.26 × Lean body weight )
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


