Disorders of Hemostasis II
![]()
INTRODUCTION
In addition to thrombocytopenia (Chapter 19) and primary deficiencies in the activity of coagulation proteins (Chapter 20), disseminated intravascular coagulation (DIC), von Willebrand disease, and qualitative abnormalities of platelets can also result in bleeding.
![]()
DISSEMINATED INTRAVASCULAR COAGULATION
Although it frequently manifests as bleeding, DIC begins as a result of an uncontrolled local or systemic activation of coagulation due to an underlying disorder. DIC may be acute or chronic, limited or diffuse, and accompanied by hemorrhage or (less commonly) thrombosis. Conditions that are associated with DIC are listed in Table 21.1.
Pathophysiology
The inciting events are numerous but generally involve either overwhelming release of tissue factor (see Chapter 20) by cellular, vascular, or hypoxemic injury, or the presence of endogenously or exogenously derived procoagulant molecules (bacterial lipopolysaccharide proteins produced by neoplastic cells).1 As coagulation is inappropriately and systemically activated, clotting factors and platelets are consumed, leading to bleeding. If activation of coagulation is chronic and low grade, however, clotting factors and platelets may be replenished and hypercoagulability may occur, manifesting as thrombosis (as in Trousseau syndrome).
Presentation
The appearance of DIC always indicates a serious underlying condition. A typical presentation of DIC involves a patient who has been hospitalized due to another disorder (Table 21.1) when unexplained bleeding and/or abnormalities in routine coagulation are observed.
![]() The hemorrhage of DIC is typically diffuse and may involve bleeding at sites of surgical incisions or vascular access catheters, as well as urinary, gastrointestinal, pulmonary, central nervous system, or cutaneous hemorrhage. Acral cyanosis and petechial and ecchymotic lesions may also occur. Widespread DIC-associated truncal and extremity bruising (Purpura fulminans) usually is limited to children or follows a viral infection. 2
The hemorrhage of DIC is typically diffuse and may involve bleeding at sites of surgical incisions or vascular access catheters, as well as urinary, gastrointestinal, pulmonary, central nervous system, or cutaneous hemorrhage. Acral cyanosis and petechial and ecchymotic lesions may also occur. Widespread DIC-associated truncal and extremity bruising (Purpura fulminans) usually is limited to children or follows a viral infection. 2
![]() Severe systemic DIC may lead to widespread tissue hypoxia and multiorgan dysfunction; hepatic, neurologic, cardiac, renal impairment may occur. The development of multiorgan dysfunction is associated with a high mortality rate.
Severe systemic DIC may lead to widespread tissue hypoxia and multiorgan dysfunction; hepatic, neurologic, cardiac, renal impairment may occur. The development of multiorgan dysfunction is associated with a high mortality rate.
Diagnosis
Typically, acute DIC is suspected when a patient with a predisposing condition (Table 21.1) develops bleeding or thrombosis and/or a perturbation in laboratory tests indicative of DIC. DIC is a dynamic condition, especially in the acutely ill patient; considerable variation in laboratory markers from timepoint to timepoint is possible, and analysis of trends rather than isolated values is crucial. Laboratory parameters may show the following:
![]() Increased (prolonged) activated partial thromboplastin time (aPTT), prothrombin time (PT), or thrombin time—due to consumption of clotting factors and/or fibrinogen (most patients).
Increased (prolonged) activated partial thromboplastin time (aPTT), prothrombin time (PT), or thrombin time—due to consumption of clotting factors and/or fibrinogen (most patients).
![]() Decreased fibrinogen (compared to baseline)*—due to consumption of fibrinogen.
Decreased fibrinogen (compared to baseline)*—due to consumption of fibrinogen.
![]() Increased products of fibrinogen and fibrin degradation (FDPs; D-dimer assay)—due to plasminmediated cleavage of fibrinogen and fibrin. The D-dimer assay measures fibrin products that have been cross-linked by activated factor XIII.
Increased products of fibrinogen and fibrin degradation (FDPs; D-dimer assay)—due to plasminmediated cleavage of fibrinogen and fibrin. The D-dimer assay measures fibrin products that have been cross-linked by activated factor XIII.
![]() Decreased platelet count (compared to baseline)*—due to clearance resulting from activation and aggregation at the sites of local prothrombotic reactions (most patients). DIC rarely produces a platelet count less than 20,000/μL. Patients with thrombosis and chronic DIC due to malignancy may have a normal or even elevated platelet count.
Decreased platelet count (compared to baseline)*—due to clearance resulting from activation and aggregation at the sites of local prothrombotic reactions (most patients). DIC rarely produces a platelet count less than 20,000/μL. Patients with thrombosis and chronic DIC due to malignancy may have a normal or even elevated platelet count.
![]() Fragmented red cells (schistocytes) on peripheral blood smear—due to microvascular hemolysis (25%–50% of patients with DIC).
Fragmented red cells (schistocytes) on peripheral blood smear—due to microvascular hemolysis (25%–50% of patients with DIC).
Treatment
The clinical and laboratory manifestations of DIC are expected to resolve with correction of the inciting disorder. This might entail effective administration of antimicrobials to a patient with sepsis, treatment of malignancy, surgery to repair an aneurysmal dilatation, removal of conceptus and placenta, or another intervention as dictated by the clinical scenario. If the DIC is severe enough to have eventuated in multiorgan dysfunction, management in an intensive care unit is required.
![]() Blood products should not be administered to patients with acute DIC unless clinically significant bleeding is present or if the risk of bleeding is felt to be high (as with thrombocytopenia in a patient who has sustained major trauma); there is, however, no reason to withhold blood products for fear of “fueling the fire.”
Blood products should not be administered to patients with acute DIC unless clinically significant bleeding is present or if the risk of bleeding is felt to be high (as with thrombocytopenia in a patient who has sustained major trauma); there is, however, no reason to withhold blood products for fear of “fueling the fire.”
![]() If bleeding is present, platelet transfusions may be administered to stop clinical bleeding; a target platelet count of 20,000 to 30,000/μL (most cases) or >50,000/μL (intracranial or life-threatening hemorrhage) is reasonable. Higher target ranges may be desired for patients who are to undergo invasive procedures such as major surgery, but the consumptive process may make achieving the goal difficult.
If bleeding is present, platelet transfusions may be administered to stop clinical bleeding; a target platelet count of 20,000 to 30,000/μL (most cases) or >50,000/μL (intracranial or life-threatening hemorrhage) is reasonable. Higher target ranges may be desired for patients who are to undergo invasive procedures such as major surgery, but the consumptive process may make achieving the goal difficult.
![]() Cryoprecipitate may be administered for bleeding in the setting of fibrinogen levels that are consistently less than 80 to 100 mg/dL. Fresh frozen plasma (FFP) should be given only to patients with significant bleeding and a prolonged PT and aPTT.
Cryoprecipitate may be administered for bleeding in the setting of fibrinogen levels that are consistently less than 80 to 100 mg/dL. Fresh frozen plasma (FFP) should be given only to patients with significant bleeding and a prolonged PT and aPTT.
![]() Due to its potential to exacerbate hemorrhage, heparin should be considered in acute DIC only in cases of bleeding, when DIC is ongoing despite appropriate treatment (blood product infusion). Heparin should not be administered unless the platelet count can be supported to 50,000/μL or higher and there is no central nervous system or diffuse gastrointestinal bleeding. If heparin is to be used, a low-dose infusion (6–10 U/kg/h) with no bolus dose is recommended. An improving platelet count and fibrinogen concentration signifies that the treatment is effective. Heparin is contraindicated in patients with placental abruption or other obstetric conditions that will require surgical management, because the anticoagulation is likely to complicate the curative treatment.
Due to its potential to exacerbate hemorrhage, heparin should be considered in acute DIC only in cases of bleeding, when DIC is ongoing despite appropriate treatment (blood product infusion). Heparin should not be administered unless the platelet count can be supported to 50,000/μL or higher and there is no central nervous system or diffuse gastrointestinal bleeding. If heparin is to be used, a low-dose infusion (6–10 U/kg/h) with no bolus dose is recommended. An improving platelet count and fibrinogen concentration signifies that the treatment is effective. Heparin is contraindicated in patients with placental abruption or other obstetric conditions that will require surgical management, because the anticoagulation is likely to complicate the curative treatment.
![]() Fibrinolysis inhibitors may have a role in patients with profuse bleeding who have failed to respond to other management, in whom FDPs are felt to be inhibiting platelets.
Fibrinolysis inhibitors may have a role in patients with profuse bleeding who have failed to respond to other management, in whom FDPs are felt to be inhibiting platelets.
![]() Due to questionable efficacy and worsened bleeding in some subjects, activated protein C concentrate (APC, drotrecogin alfa) is no longer recommended for patients with severe sepsis and DIC.3 Use of antithrombin concentrates is controversial. 4
Due to questionable efficacy and worsened bleeding in some subjects, activated protein C concentrate (APC, drotrecogin alfa) is no longer recommended for patients with severe sepsis and DIC.3 Use of antithrombin concentrates is controversial. 4
![]() Laboratory parameters (PT, PTT, fibrinogen, and platelet count) should be monitored at least every 6 hours in the acutely ill patient with DIC, and clinical bleeding should be followed to assess efficacy of therapeutic measures.
Laboratory parameters (PT, PTT, fibrinogen, and platelet count) should be monitored at least every 6 hours in the acutely ill patient with DIC, and clinical bleeding should be followed to assess efficacy of therapeutic measures.
HELLP syndrome (hemolysis, elevated liver enzymes, and low platelets) affects women in the peri-partum period and produces clinically significant hemolytic anemia, hepatocellular injury, and low platelets. It may be difficult initially to distinguish the disorder from thrombotic thrombocytopenia purpura (TTP). Hepatic dysfunction (leading to elevated transaminases) may distinguish the diagnosis from TTP, which also may complicate pregnancy (see Chapter 19). Introduction of placental proteins into the maternal circulation has been thought to be etiologic; potential biomarkers have been identified.5 Gross hemoglobinuria with renal dysfunction and hypotension is common; the mortality rate is high. Management must include evacuation of the uterus, either by delivery of a term or near-term infant, or by dilatation and curettage to remove retained placental or fetal tissue.
Acute promyelocytic leukemia (APL) is frequently accompanied by DIC, potentially due to procoagulant molecules (tissue factor and others) contained within circulating promyelocytes. Bleeding commonly occurs in the lungs and brain and is frequently fatal. In addition to the appropriate use of blood products (FFP, cryoprecipitate, platelets) upon detection of APL-associated DIC, emergent initiation of treatment with all-trans-retinoic acid (ATRA) is recommended (see Chapter 11).6
Trousseau syndrome is a form of chronic DIC in which recurrent episodes of venous thromboembolism (VTE) complicate an underlying malignancy, especially adenocarcinomas. Experience in the management of the disorder has suggested that anticoagulation with warfarin is not effective in preventing further VTE; instead, subcutaneous low molecular weight heparin in therapeutic doses is usually necessary to prevent recurrence of thromboembolism (see Chapter 23).7
![]()
VON WILLEBRAND DISEASE
Epidemiology
von Willebrand disease (VWD) is the most common inherited bleeding disorder.8
Pathophysiology and Classification
Von Willebrand factor (VWF) is an extremely large multimeric glycoprotein that is synthesized in endothelial cells and megakaryocytes. Binding of VWF to its receptor, platelet glycoprotein Ib (GPIb), tethers platelets to one another and to the subendothelial collagen matrix, localizing them to the site of injury. This interaction is especially important to assure primary hemostasis in vessels such as arterioles, where a “high shear” state is present (Fig. 21.1). VWF also binds factor VIII (FVIII) in the circulation, protecting it from clearance.
![]() Type 1 (quantitative defect in VWF) includes approximately 75% to 80% of patients, the majority of whom do not have an identified causal mutation in the VWF gene, which is located on chromosome 12. Patients may have mild or moderate bleeding. Autosomal dominant inheritance is typical.
Type 1 (quantitative defect in VWF) includes approximately 75% to 80% of patients, the majority of whom do not have an identified causal mutation in the VWF gene, which is located on chromosome 12. Patients may have mild or moderate bleeding. Autosomal dominant inheritance is typical.
![]() Type 2 (qualitative defect in VWF) includes four subtypes; patients usually have moderate bleeding symptoms and present before adulthood. Type 2A (10% to 15% of VWD) involves mutations in VWF that cause either a defect in intracellular transport (2A, type 1) or render the molecule more susceptible to proteolysis (2A, type 2). Laboratory testing (Table 21.2) typically shows a marked decrease in VWF activity relative to antigen (a ratio of ≤0.6 is typical). Type 2B (5% of VWD) mutations result in an abnormal structure in the binding site for platelet GPIb (A1 domain of VWF), and are responsible for a “gain-of-function” defect that allows spontaneous binding of the abnormal VWF to platelets in the circulation. Patients typically have thrombocytopenia due to removal of VWF-bound platelet aggregates. The ristocetin-induced platelet aggregation (RIPA) (Table 21.2) shows an increase in platelet aggregation to low concentrations of ristocetin.† Type 2N (uncommon) features mutations in VWF that decrease its ability to bind and protect FVIII from clearance, resulting in decreased FVIII levels in the plasma and a phenotype similar to hemophilia A. Soft tissue and joint bleeding are common. The presence of affected females in the family is an important clue to consider this diagnosis. Laboratory studies show decreased FVIII (2% to 10%), and normal VWF function and antigen. Type 2M (very uncommon) results from mutations affecting the A1 domain in an area different from those mutations in type 2B. These result in decreased binding of platelets to VWF.
Type 2 (qualitative defect in VWF) includes four subtypes; patients usually have moderate bleeding symptoms and present before adulthood. Type 2A (10% to 15% of VWD) involves mutations in VWF that cause either a defect in intracellular transport (2A, type 1) or render the molecule more susceptible to proteolysis (2A, type 2). Laboratory testing (Table 21.2) typically shows a marked decrease in VWF activity relative to antigen (a ratio of ≤0.6 is typical). Type 2B (5% of VWD) mutations result in an abnormal structure in the binding site for platelet GPIb (A1 domain of VWF), and are responsible for a “gain-of-function” defect that allows spontaneous binding of the abnormal VWF to platelets in the circulation. Patients typically have thrombocytopenia due to removal of VWF-bound platelet aggregates. The ristocetin-induced platelet aggregation (RIPA) (Table 21.2) shows an increase in platelet aggregation to low concentrations of ristocetin.† Type 2N (uncommon) features mutations in VWF that decrease its ability to bind and protect FVIII from clearance, resulting in decreased FVIII levels in the plasma and a phenotype similar to hemophilia A. Soft tissue and joint bleeding are common. The presence of affected females in the family is an important clue to consider this diagnosis. Laboratory studies show decreased FVIII (2% to 10%), and normal VWF function and antigen. Type 2M (very uncommon) results from mutations affecting the A1 domain in an area different from those mutations in type 2B. These result in decreased binding of platelets to VWF.
![]() Type 3 VWD (rare) is caused by a variety of mutations of the VWF molecule, including larger deletions; patients may be homozygous for a given mutation or double heterozygotes. Severe bleeding manifests in childhood. FVIII is usually about 5%, and VWF levels usually are too low to detect.
Type 3 VWD (rare) is caused by a variety of mutations of the VWF molecule, including larger deletions; patients may be homozygous for a given mutation or double heterozygotes. Severe bleeding manifests in childhood. FVIII is usually about 5%, and VWF levels usually are too low to detect.
Presentation
Bleeding symptoms usually involve mucous membranes. Epistaxis, oral bleeding, menorrhagia, and gastrointestinal bleeding are common. Individuals with marked abnormalities of VWF usually present earlier in life with bleeding at the time of mucous membrane-related procedures (tooth extractions, tonsillectomy), or at menarche.
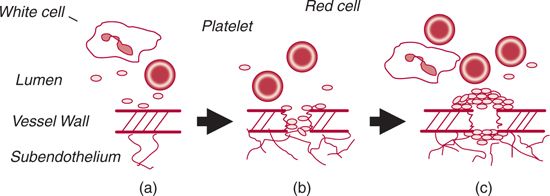
FIGURE 21.1. Primary hemostasis. A. Normal conditions. Under physiologic conditions, platelets do not interact with the endothelium. B. Adhesion. Upon disruption of the blood vessel wall, subendothelial collagen and fibronectin are exposed, leading to platelet adhesion. In the arterial/arteriolar circulation, subendothelial von Willebrand factor (VWF) assists in the adherence of platelets to the site of injury via binding to the platelet glycoprotein (GP) Ib receptor. C. Aggregation. Tissue factor interacts with factor VIIa, present locally, to catalyze the formation of thrombin.Thrombin, collagen, and other molecules bind to receptors on the platelet membrane, leading to platelet activation. Fibrinogen cross-links platelets via their GPIIb/IIIa receptors, promoting formation of an occlusive plug that prevents additional blood loss through the break in the vessel wall.VWF also bridges between platelets, via their GPIb and GPIIb/IIIa receptors.

Diagnosis
The diagnosis of VWD is based on a typical history of bleeding (i.e., mucous membrane-related) and confirmatory laboratory testing.9 The diagnosis can be difficult, given the large number of individuals (estimated up to 1% of the population) whose VWF levels fall below the laboratory reference range, many of whom do not experience abnormal bleeding.
![]() The personal and family history of bleeding should be carefully documented.
The personal and family history of bleeding should be carefully documented.
![]() A complete blood count and routine coagulation studies should be performed, to exclude other diagnoses and assess for anemia.
A complete blood count and routine coagulation studies should be performed, to exclude other diagnoses and assess for anemia.
![]() Initial testing for VWD (Table 21.2). A VWF antigen level (by ELISA) and a VWF activity (by ristocetin cofactor assay) should be performed. The latter involves addition of ristocetin at 1.2 mg/mL to a mixture of patient plasma (the VWF source) and washed normal platelets. Ristocetin binds VWF, allowing it to bind GPIb on the platelet membrane, causing platelet aggregation. The factor VIII activity may be abnormal.
Initial testing for VWD (Table 21.2). A VWF antigen level (by ELISA) and a VWF activity (by ristocetin cofactor assay) should be performed. The latter involves addition of ristocetin at 1.2 mg/mL to a mixture of patient plasma (the VWF source) and washed normal platelets. Ristocetin binds VWF, allowing it to bind GPIb on the platelet membrane, causing platelet aggregation. The factor VIII activity may be abnormal.
![]() Secondary testing. A VWF multimer study detecting the distribution of multimers is performed once a diagnosis of VWD has been made, to assess for type 2 VWD.
Secondary testing. A VWF multimer study detecting the distribution of multimers is performed once a diagnosis of VWD has been made, to assess for type 2 VWD.
![]() VWF levels <30% are regarded by most clinicians as diagnostic of VWD.
VWF levels <30% are regarded by most clinicians as diagnostic of VWD.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


